fetching data ... 
Background: Brown syndrome is a rare ocular motility disorder which has been reported in JRA, RA and SLE but never in a patient with scleromyositis.
Objectives: To report the first case of Brown syndrome in a patient with scleromyositis and increase awareness of this condition.
Methods: A case report and discussion.
Results: The patient was diagnosed with scleromyositis, at the age of 34, after presenting with arthralgia, sclerodactyly, skin pigmentation, Raynaud’s phenomenon, mild muscle weakness and dyspnoea. His labs were CRP 47 mg/L, CK 868 IU/L, ANA strongly positive; anticentromere Ab negative and Anti-PM/Scl-75 and Anti- PM/Scl-100 Ab positive. HRCT chest showed extensive pulmonary fibrosis with lower lobe honeycombing. TLCO was 3.98 (33% of predicted).
He was initially managed with high dose steroids and pulsed IV cyclophosphamide with azathioprine for maintenance therapy. His lung disease stabilised and myositis resolved but he continued to develop calcinosis cutis so was switched to 6 monthly IV rituximab.
6 years later, he developed morning headaches with intermittent diplopia, described as double vision in vertical gaze with one image being above the other. Episodes lasting 10 minutes to 2 hours. Examination showed normal visual acuity and fundoscopy, no peripheral or eye muscle weakness.
Investigations to exclude myasthenia gravis, cerebral vasculitis and atypical infection were organised (MRI, AChR antibody, lumbar puncture, MRA) and were normal.
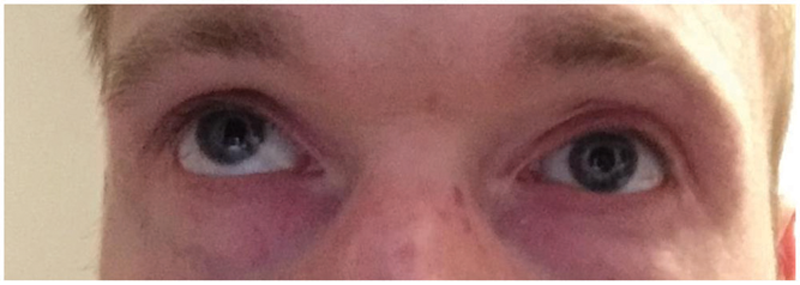
Because of intermittent nature of his episodes, his eye examination was always normal but he captured images in disconjugate gaze with right eye looking upwards and outwards when trying to look straight (
He continues to have recurrent episodes despite immunosuppression but prednisolone 20mg daily for 1-2 days at onset of each attack causes rapid resolution of symptoms.
Right eye looking upwards and outwards when trying to look straight
Conclusion: Scleromyositis is an overlap syndrome of scleroderma and dermatomyositis. Muscle involvement is mild and clinical presentation can be variable. The PM/Scl antibodies are highly characteristic of the syndrome. (1)
Brown syndrome is an ocular motility disorder, first described in 1950, characterized by the inability to fully elevate the affected eye in adduction due to pathology of the superior oblique tendon sheath. (2)
It can be congenital or acquired, viz, trauma, surgery or sinusitis and also been described in RA, JIA and SLE. (3)
If superior oblique tendon cannot relax or slide freely through the trochlea then the affected eye cannot depress completely, leading to diplopia on upward gaze. (4) In inflammatory disease it is thought that swelling of the posterior part of the superior oblique tendon or tenosynovitis are likely causes of the tendon sheath abnormality. (4) This is likely to be the case in this patient because his symptoms are recurrent, respond to steroids and tend to occur more towards the end of rituximab cycles.
Recognition of this syndrome is important because invasive investigations can be avoided. Also, intermittent diplopia in a patient with autoimmune disease is suggestive of myasthenia gravis which maybe incorrectly diagnosed.
Finally, this case demonstrates the syndrome can be easily managed with short courses of oral steroids, although patients who are already on immunosuppressant treatment may need this in addition.
REFERENCES:
[1]Török L, Dankó K, Cserni G, Szûcs G. PM-SCL autoantibody positive scleroderma with polymyositis (mechanic’s hand: clinical aid in the diagnosis). JEADV 2004; 18: 356–359
[2]Brown H W. Congenital structural muscle anomalies. In:Alien J H, ed. Strabismus ophthalmic symposium I. St Louis:CV Mosby, 1950: 205-6.
[3]Cooper C, Kirwan JR, McGill NW, Dieppe PA. Brown’s syndrome: an unusual ocular complication of rheumatoid arthritis. Ann Rheum Dis 1990; 49:188-9.
[4]Sandford-Smith JH. Superior oblique tendon syndrome and its relationship to stenosing tenosynovitis. Br JOphthalmol 1973; 57:859-65.
Disclosure of Interests: None declared