fetching data ... 
Background: Systemic lupus erythematosus (SLE) is a complex autoimmune disease with unknown etiology involving multiple immune cells and has diverse clinical phenotypes. This heterogeneous nature has hampered a better understanding of SLE pathogenesis and the development of effective therapeutic agents. While recent single-cell RNA sequencing studies of SLE identified several important cell subpopulations, they were limited by sparse expression information at single-cell level and small sample sizes.
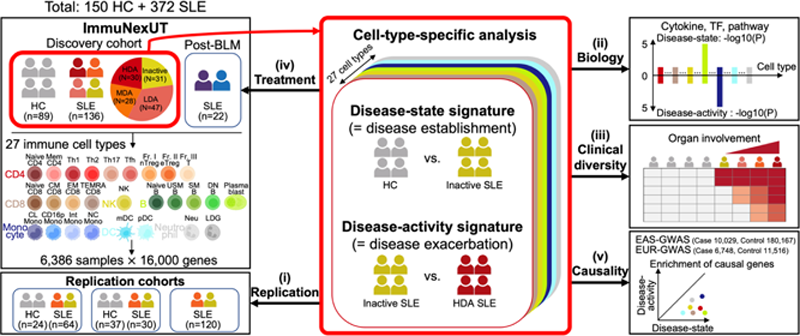
Objectives: This study aimed to elucidate the dysregulated gene expression pattern linked to multiple clinical statuses of SLE with a fine cellular resolution and higher sensitivity. We also attempted to resolve a complex interaction between risk variants and the transcriptome dysregulation seen in SLE patients.
Methods: We conducted a large-scale bulk transcriptome study of 6,386 RNA-sequencing data including 27 purified immune cell types in peripheral blood from 136 SLE and 89 healthy donors in the Immune Cell Gene Expression Atlas from the University of Tokyo (ImmuNexUT) cohort 1 . At enrollment, SLE patients had diverse clinical manifestations (disease activity, organ involvement and treatment profiles) and 22 patients were re-evaluated after belimumab treatment.
Results: We first profiled two distinct cell-type-specific transcriptomic signatures: disease-state and disease-activity signatures, reflecting disease establishment and exacerbation, respectively.
After confirming the high replicability of both signatures in independent cohorts, we identified candidates of biological processes unique to each signature: e.g., upregulated E2F transcriptional activity in Th1, CD8+ memory T-lineage and NK cells, and dynamic increase of IL21 and CXCL13 in Th1 cells in an active phase of SLE. Pathway analysis highlighted the importance of immunometabolic process for SLE (e.g., oxidative phosphorylation) in cell-type-specific resolution.
Moreover, we demonstrated cell-type-specific contributions to diverse organ involvement, e.g., Th1 for mucocutaneous, monocyte-lineage cells for musculoskeletal, neutrophil-lineage cells for renal activity, respectively.
We also observed the strong associations of disease-activity signatures with treatment effect: (i) belimumab suppressed activity signatures from B-lineage cells, especially in good responders and (ii) mycophenolate mofetil substantially suppressed activity signatures from plasmablast, Th1, and central memory CD8 cells.
However, through stratified LD score regression using large-scale SLE-GWASs, we revealed that disease-activity signatures were less enriched around SLE risk variants than disease-state signatures. Consistent with this result, the directions of SLE risk alleles’ expression quantitative trait locus (eQTL) effects were significantly concordant with the directions of disease-state signatures, but not with those of activity signatures. These findings suggested that the current genetic case-control studies may not well capture clinically vital biology linked to drug target discovery for SLE. Meanwhile, we also detected some examples of activity signatures that might contribute to the disease risk by modulating risk allele’s eQTL effects.
Conclusion: We identified comprehensive gene signatures reflecting the establishment and exacerbation of SLE, which provide essential foundations for future genomic, genetic, and clinical studies.
REFERENCES:
[1]Ota, M. et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 2021;184:3006-21.e17.
Acknowledgements: This study was supported by Chugai Pharmaceutical Co., Ltd., Tokyo, Japan; the Ministry of Education, Culture, Sports; and the Japan Agency for Medical Research and Development (AMED) (JP21tm0424221 and JP21zf0127004).
Disclosure of Interests: Masahiro Nakano: None declared, Mineto Ota Grant/research support from: Mineto Ota belongs to the Social Cooperation Program, Department of functional genomics and immunological diseases, supported by Chugai Pharmaceutical., Yusuke Takeshima Grant/research support from: Yusuke Takeshima belonged to the Social Cooperation Program, Department of functional genomics and immunological diseases, supported by Chugai Pharmaceutical., Yukiko Iwasaki: None declared, Hiroaki Hatano: None declared, Yasuo Nagafuchi Grant/research support from: Yasuo Nagafuchi belongs to the Social Cooperation Program, Department of functional genomics and immunological diseases, supported by Chugai Pharmaceutical., Kwangwoo Kim: None declared, So-Young Bang: None declared, Hye Soon Lee: None declared, Hirofumi Shoda: None declared, Xuejun Zhang: None declared, Sang-Cheol Bae: None declared, Chikashi Terao: None declared, Kazuhiko Yamamoto: None declared, Tomohisa Okamura Grant/research support from: Tomohisa Okamura belongs to the Social Cooperation Program, Department of functional genomics and immunological diseases, supported by Chugai Pharmaceutical., Kazuyoshi Ishigaki: None declared, Keishi Fujio Speakers bureau: Keishi Fujio receives speaker fees from Chugai Pharmaceutical., Consultant of: Keishi Fujio receives consulting honoraria from Chugai Pharmaceutical., Grant/research support from: Keishi Fujio receives research support from Chugai Pharmaceutical.