fetching data ... 
Background: Patients with systemic lupus erythematosus (SLE) display an aberrant DNA methylation (DNAm) pattern with a predominant interferon signature. However, the DNAm patterns in purified immune cell populations are not well described.
Objectives: To examine genome-wide DNAm changes in sorted CD4 + T cells, monocytes, granulocytes and B cells in SLE patients compared to healthy controls (HC).
Methods: Genome-wide analysis was performed in 20 SLE patients with active lupus nephritis (LN) and 20 age-, sex- and ethnicity-matched HCs. Peripheral blood was processed using gradient density centrifugation for the granulocyte fraction, and CD4 + T-cells, monocytes and B cells were isolated from the mononuclear cell fraction using a RoboSep device (Stemcell Technologies). DNA was extracted using the DNA/RNA/miRNA Universal kit (Qiagen). Genome-wide DNAm was evaluated using Infinium MethylationEPIC BeadChips (Illumina Inc.). Top differentially methylated CpG sites (DMC) were validated using pyrosequencing, and further analysed in follow-up samples, as well as extended to another 36 SLE patients with inactive LN, 36 SLE with never-LN and 20 HCs. Patients and HCs were compared using a paired t -test stratified by cell type. A false discovery rate (FDR) <0.05 was considered statistically significant. Clustering of non-gene annotated CpGs was defined as distance <5000 bp with proximity to neighbouring genes <1x10 5 bp, identified through mapping of Entrez Gene Identifiers.
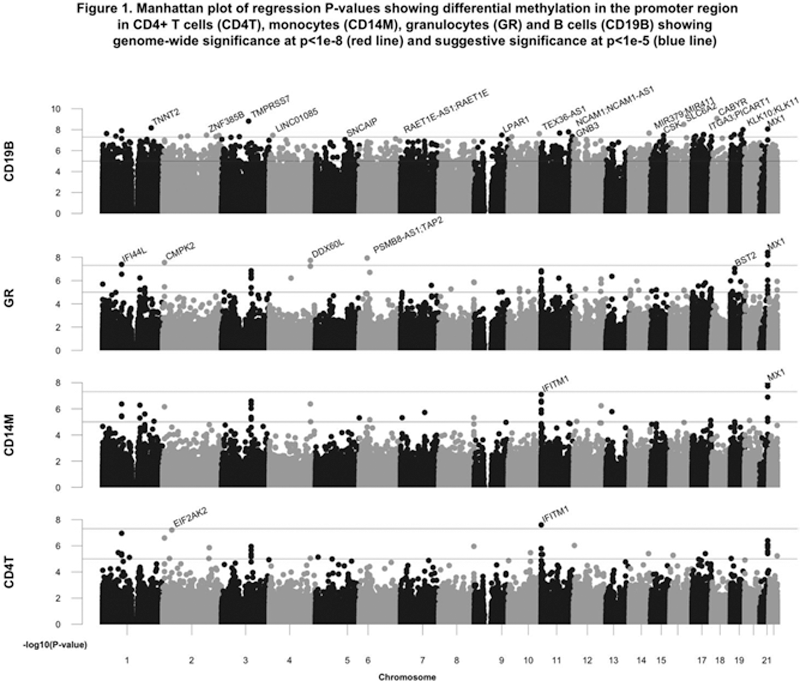
Results: Overall, SLE patients with active LN compared to HCs exhibited DMCs in 22, 46, 312 and 78,068 probes in CD4
+
T cells, monocytes, granulocytes and B cells, respectively. In CD4
+
T cells, monocytes and granulocytes, the majority of DMCs were hypomethylated and related to interferon-regulated genes. In B cells, the majority of DMCs were hypermethylated with 1360 genes in the promoter region restricted to differential methylation >10% and ≥2 DMCs, of which 1087 were hypermethylated. Of these, the most significantly hypermethylated CpGs in the promoter region included
CABYR, TMPRSS7, SLC6A2, KLK10/KLK11, WIZ, LPAR1, ZNF385B
and
IL1R1
(
Pyrosequencing confirmed B cell hypermethylation in CXCR5, DDR1, MTA3, RAB30 and TRAF5, and in non-coding clusters located on chromosome 3 upstream of FGF12 and chromosome 22 downstream of XPNPEP3 and RBX1 . Hypermethylation in CXCR5, DDR1, MTA3, RAB30 and hypomethylation in IFI44L, LGALS3BP and PARP9 was stable at follow-up as well as in patients with inactive and never-LN. Patients in the active LN group exhibited significantly more pronounced hypomethylation in PLSCR1 in CD4 + T-cells, granulocytes and B-cells compared to the other patient groups. Greater β2 microglobulin, anti-DNA antibodies and disease activity were significantly associated with greater B cell CXCR5 hypermethylation (p<0.001), IFI44L hypomethylation in all but CD4 + T cells , LGALS3BP hypomethylation in granulocytes (p<0.0001) , PARP9 hypomethylation in all but B cells and PLSCR1 hypomethylation in all but monocytes. Proteinuria was related to PLSCR1 hypomethylation in all but monocytes, as well as hypermethylation at the non-coding cluster in chromosome 3 (p=0.05) and MTA3 (p=0.03) in B cells.
Conclusion: B cell hypermethylation in SLE patients is widespread, and may indicate a novel mechanism for SLE pathogenesis. Differential methylation of several interferon-regulated genes may be associated with disease activity.
Disclosure of Interests: None declared