fetching data ... 
Background: Antiphospholipid syndrome (APS) is an autoimmune thrombophilia associated with antibodies against phospholipid binding plasma proteins, mainly β2glycoprotein-I (B2GPI), prothrombin (PT) and annexin[1]. APS is expressed mainly as arterial or venous thrombosis and pregnancy complications. APS may occur as isolated disease (primary APS) or secondary to another autoimmune disease, mainly systemic lupus erythematosus (SLE)[2].
Objectives: To describe the clinical and serological characteristics of a cohort of Greek patients with Primary APS, to check the performance of the 2023 ACR/EULAR APS classification criteria in this cohort and to highlight the non-criteria clinical phenotypes.
Methods: A retrospective study in a Greek cohort of Primary APS. Patient demographics, clinical and serological features were recorded. Comparisons are made between classified and non-classified APS patients by using chi-square and one way ANOVA tests. Significance was set at p < 0.05.
Results: Sixty-eight patients with primary APS were included. Median age was 55 years (21-80), 69% female and 29% were active smokers (mean packyears: 32 ± 12). The most often implicated antiphospholipid antibodies were ΙgM and ΙgG anti-cardiolipin (51.5% and 29.4%), anti-β2GPI IgG and IgM (25% and 41.2%) and lupus anticoagulant (48.5%). Clinical manifestations were venous thrombosis in 34 patients (50%) [most often deep venous thrombosis (n=20), pulmonary embolism (n=16) and cavernous sinus thrombosis (n=3)], arterial thrombosis in 25 patients (36.8%) [most often stroke (n=18)], and pregnancy morbidity in 18 patients (26.4%). Other manifestations included: livedo racemosa (n=3, 4.4%), APS nephropathy (n=4, 5.8%), cardiac valve disease (n=2, 2.9%), thrombocytopenia (n=19, 27.9%) and catastrophic APS (n=1, 1.4%). The following were considered as non-criteria manifestations: migraine (n=6, 8.8%), multiple-sclerosis-like brain lesions (n=6, 8.8%), skin ulcers (n=5, 7.4%), transverse myelitis (n=4, 5.9%), osteonecrosis (n=3, 4.4%), Raynaud’s phenomenon (n=2, 2.9%), malar rash (n=1, 1.4%) and sensorineural hearing loss (n=1, 1.4%).
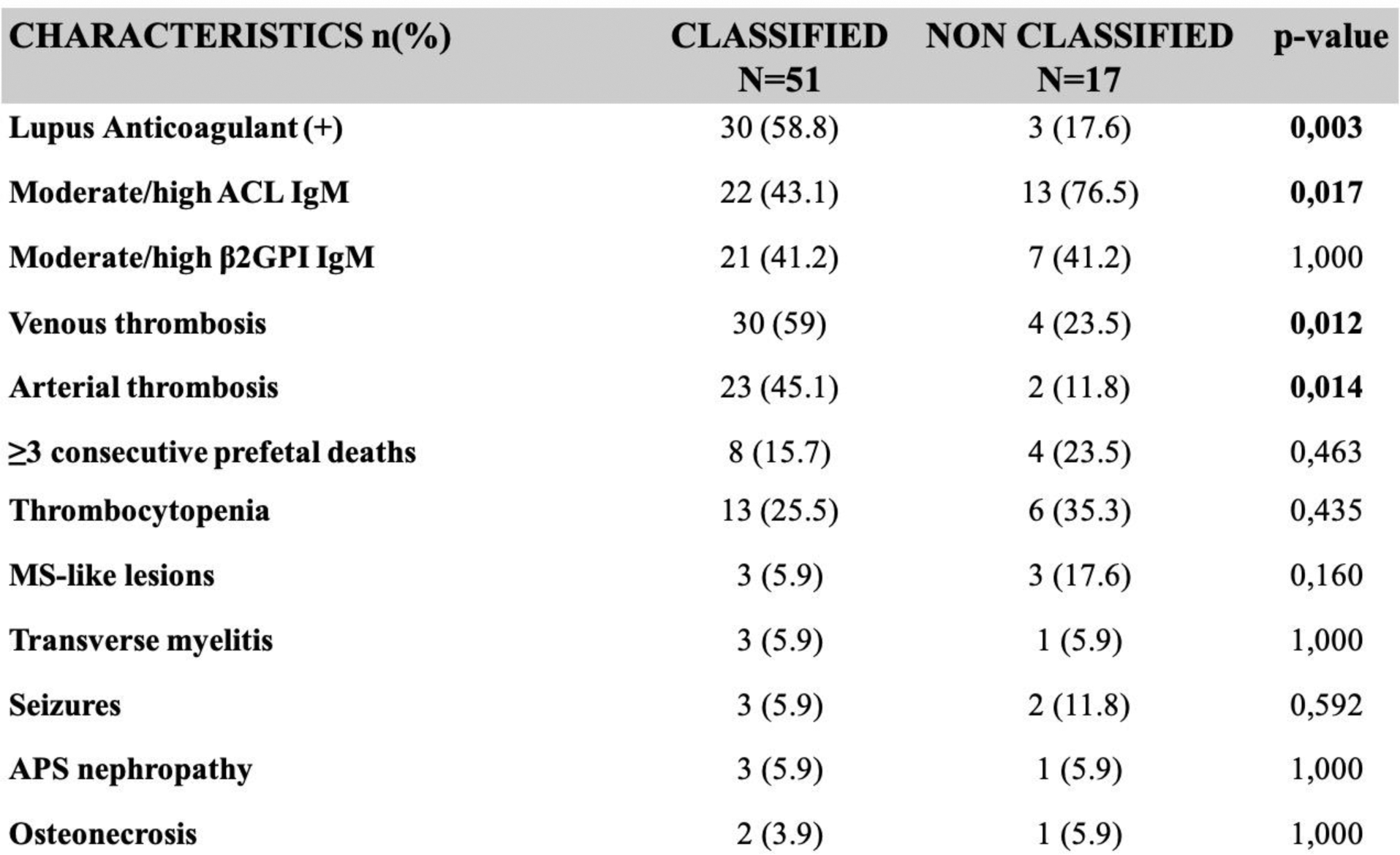
Fifty-one patients (75%) were classified as APS according to the 2023 ACR/EULAR APS Classification Criteria. The arm of non-classified patients included fewer active smokers (5.9% vs 35%, p=0.027), more patients with a moderate/high anti-cardiolipin IgM titre (76.5% vs 43.1%, p=0.017), less arterial (11.8% vs 45.1%, p=0.014) and venous thromboses (23.5% vs 59%, p=0.012). Characteristics that yielded non-significant differences included: thrombocytopenia, three or more otherwise unexplained consecutive prefetal deaths and moderate/high anti-β2GPI IgM (Figure 1).
Conclusion: Our study highlights the different clinical phenotypes of APS. In agreement with the existing literature, a portion of patients do not meet the new classification criteria. Therefore, clinicians should evaluate patients carefully, as there are no diagnostic criteria. It is up to the clinician to interpret the clinical manifestations, antibody profile and other risk factors for the diagnosis of APS.
REFERENCES: [1] Khamashta MA, Amigo MC. Antiphospholipid syndorme: overview of pathogenesis, diagnosis, and management. In: Rheumatology, 6, Hochberg MC, Silman AJ, Smolen JS, et al (Eds), Elsevier, 2015. Vol 2, p.1144.
[2] Taraborelli M, Leuenberger L, Lazzaroni MG, et al. The contribution of antiphospholipid antibodies to organ damage in systemic lupus erythematosus. Lupus 2016; 25:1365.
Comparisons between classified and non-classified Primary APS patients (2023 ACR/EULAR classification criteria)
Acknowledgements: NIL.
Disclosure of Interests: None declared.