fetching data ... 
Background: The antiphospholipid syndrome (APS) is a systemic autoimmune disease (SADs) characterized by thrombotic events, pregnancy losses, and associated specific antibodies (aPL). It can be primary or associated with other SADs. It is known that the presence of specific aPL, either individually or in combination, confers different risks for developing certain events in APS. Until now, the diagnosis was made following the classification criteria of Sapporo 1999, revised in Sydney in 2004, where specific aPL were included under certain conditions. In 2023, the criteria proposed by the American College of Rheumatology (ACR)/European Alliance of Associations for Rheumatology (EULAR) were published, which incorporated new demographic and clinical manifestations, and different aPL criteria, for a better patient classification.
Objectives: To describe the association between the different aPL in APS assessed according to both the Sydney and the new ACR/EULAR criteria and the demographic and clinical characteristics of the patients.
Methods: Retrospective study of 81 patients with two separate positive determinations for aPL at least 12 weeks apart at Hospital del Mar (Barcelona, Spain) between 2011 and 2021. Demographic, clinical, and biological data were collected. A bivariate statistical analysis was conducted in IBM® SPSS 25.0, using Chi-square or Fisher’s exact test for qualitative variables and the Student’s t-test or Mann-Whitney U test for continuous variables.
Results: Among the 81 patients in our cohort, it was observed that 62 (76.543%) were aPL asymptomatic carriers and 19 (23.46%) met any of the 2 classification criteria. Table 1 describes statistically significant differences between demographic or clinical manifestations and levels of aPL. In our cohort, it was noted that patients with lupus anticoagulant were diagnosed at an earlier age than the rest, and those with elevated levels of anticardiolipin (aCL) or beta 2 -glycoprotein I antibodies (β2GCP) for both IgM and IgG, along with those with triple aPL positivity, had a higher frequency of thrombocytopenia. Furthermore, an association was observed between the presence of aCL or β2GCP IgM and valve thickening, as well as between their high titers and fetal death. It is noteworthy the association found between high titers of β2GCP and arterial events as well as the one between having thrombotic events during antiplatelet treatment in those with triple aPL positivity. The association with other SADs was more frequent in patients with aCL or β2GCP, regardless of their titers. No statistically significant differences were found in patients with 2 concomitant aPL.
Conclusion: Different profiles of antibodies according to various immunological criteria collected in different classification criteria are associated with specific clinical manifestations. Understanding these specific associations is important to guide us in the diagnosis, management, and treatment of these patients. It is important to highlight that, in the presence of triple positivity, the risk of thrombosis should be considered despite antiplatelet treatment.
Table 1. Statistically significant relationship between antibodies associated with antiphospholid syndrome and demographic or clinical manifestations. Number of patients with positive clinical characteristic and positive aPL/ Number of patients with positive clinical characteristic and negative aPL.
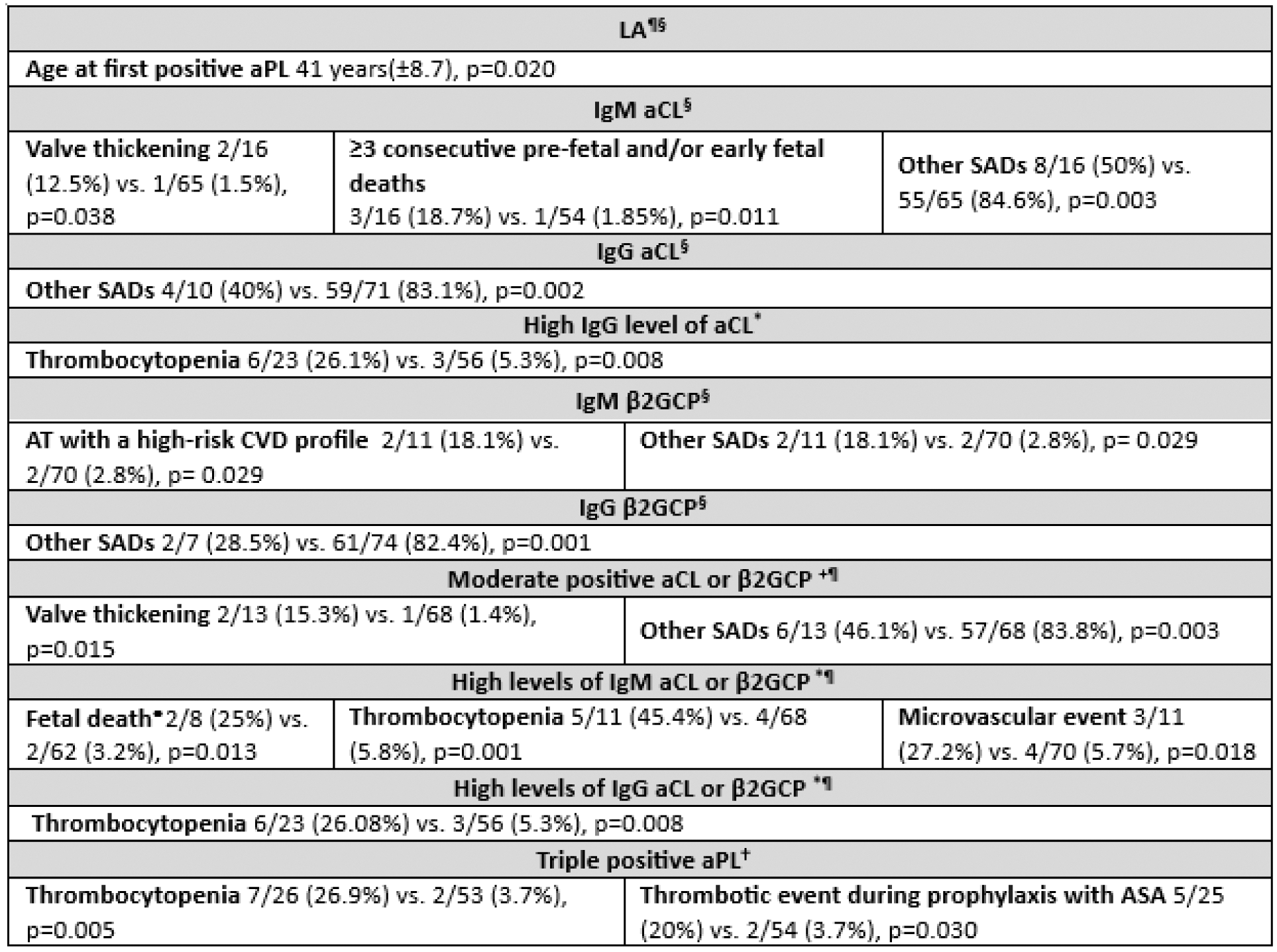
§ Criterion included in Sydney classification criteria. ¶ Criterion included in ACR/EULAR classification criteria. *High titers ≥80units. + Moderate titers ≥40-79 units. • Unexplained pregnancy loss between 10 weeks 0 days and 15 weeks 6 days gestation (early fetal death), or between 16 weeks 0 days and 34 weeks 0 days gestation. † Anti-beta 2 -glycoprotein I antibodies, anticardiolipin antibody, and lupus anticoagulant. AT: arterial thrombosis, ASA: Acetylsalicylic acid, aPL: Antiphospholipid antibodies, aCL: Anticardiolipin antibodies, β2GCP: beta 2 -glycoprotein I antibodies, CVD: cardiovascular disease, Ig: Immunoglobulin, LA: Lupus anticoagulant, SADs: systemic autoimmune diseases.
REFERENCES: NIL.
Acknowledgements: NIL.
Disclosure of Interests: None declared.