fetching data ... 
Background: Characterized by its clinical diversity and multisystem involvement, Systemic lupus erythematosus (SLE) is a complex autoimmune disease. The development of SLE is determined by a combination of both genetic and environmental factors. It is well documented that Epstein–Barr virus (EBV) infection is closely associated with SLE. However, it remains unclear which genes and immune cells are pivotal in EBV-infected SLE patients.
Objectives: We aimed to identify overlapping differentially Expressed Genes (DEGs) and immune cell dynamics in both patients with SLE and patients with EBV infection using array data. Subsequently, we aim to further analyze and verify the results of our single-cell sequencing studies.
Methods: The public microarray data sets of SLE (GSE50772 and GSE81622) or EBV infection (GSE85599 and GSE45918) were obtained from the Gene Expression Omnibus (GEO) database. Subsequently, we conducted differential gene expression analysis to identify overlapping DEGs and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was carried out based on the overlapping DEGs. Subsequently, machine learning approaches were applied to identify key genes from the overlapping DEGs. Finally, we conducted single cell RNA-sequencing in PBMCs isolated from EBV positive SLE patients (SLE-EBV+), EBV negative SLE patients (SLE-EBV-) and normal controls.
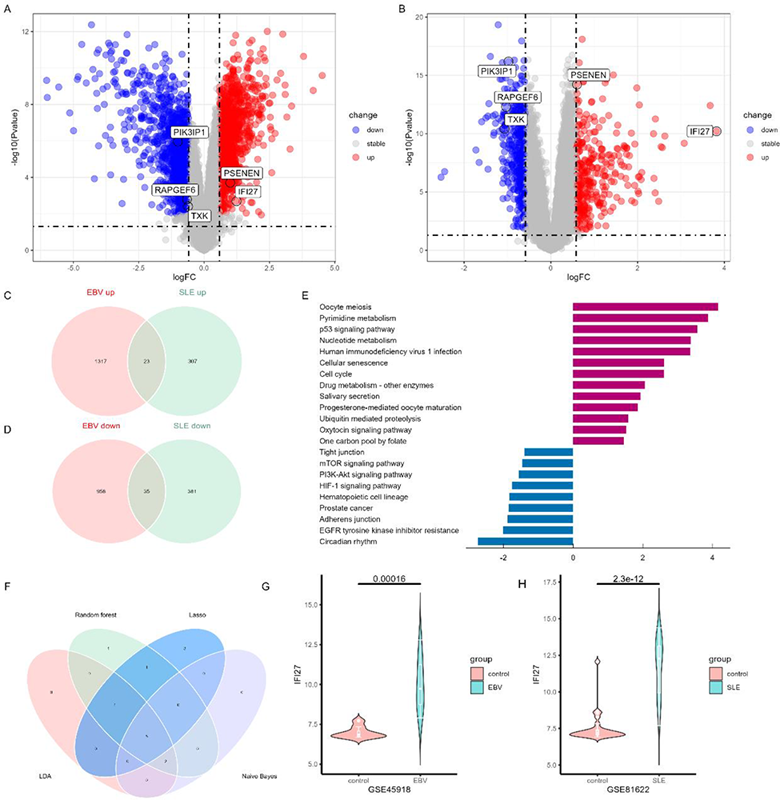
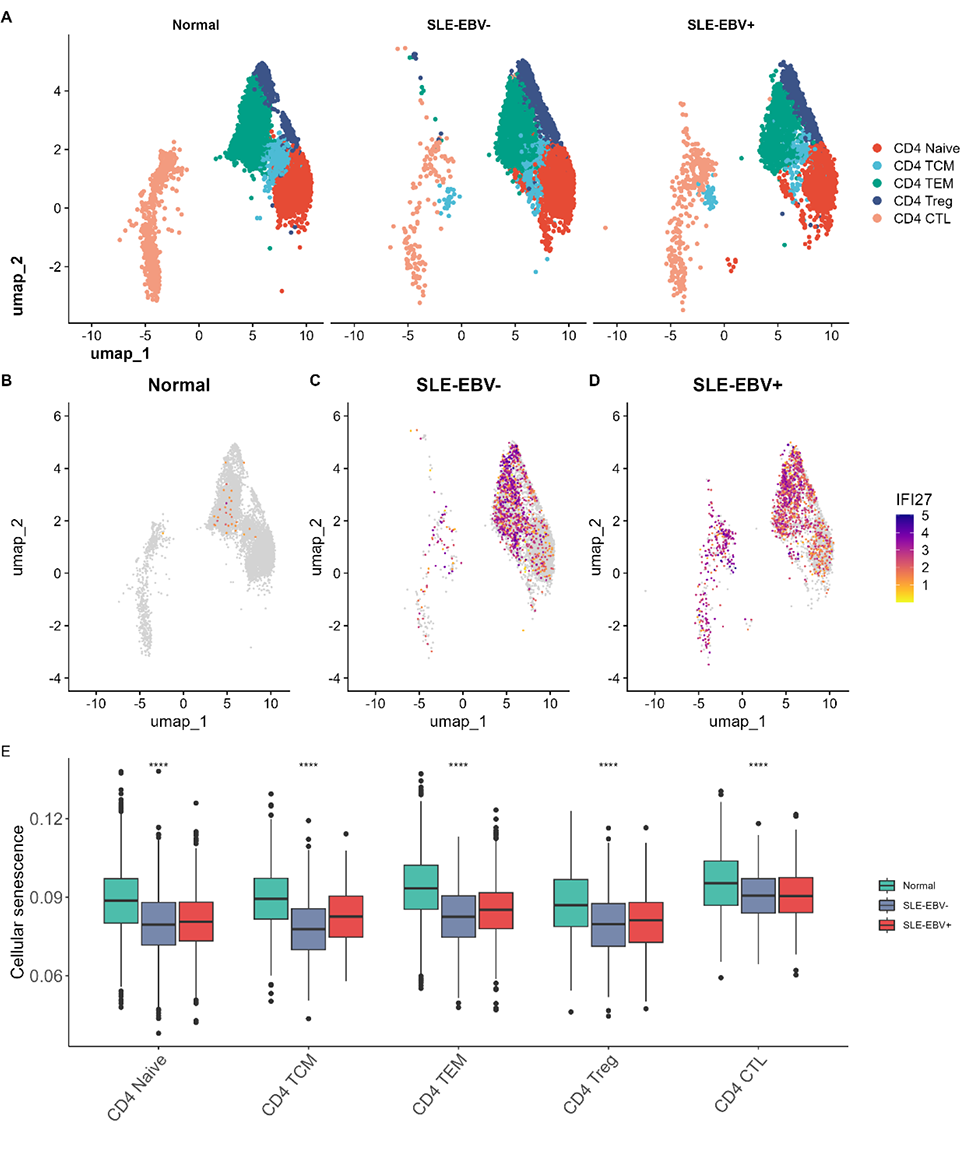
Results: In total, 58 overlapping DEGs were identified, comprising 23 up-regulated genes and 35 down-regulated genes. The most significantly enriched pathways were recognized as oocyte meiosis and circadian rhythm. In addition, we employed machine learning algorithms to select 5 genes (IFI27, TXK, RAPGEF6, PIK3IP1, PSENEN) as key genes (Figure 1). The proportion of T cells CD4 memory resting was significantly decreased in both the SLE database and the EBV infection database. In our scRNA-seq data, the expression of IFI27 was significantly different in all CD4 T cell subsets between SLE-EBV+ and SLE-EBV- patients (Figure 2). Significant differences were observed in the expression of the other four genes (TXK, RAPGEF6, PIK3IP1, PSENEN) in CD8 T cells between SLE-EBV+ and SLE-EBV- patients. TXK, RAPGEF6, and PIK3IP1 also exhibited significant differences in monocytes between SLE-EBV+ patients and EBV- patients.
Conclusion: The identification of five key genes (IFI27, TXK, RAPGEF6, PIK3IP1, and PSENEN) suggests their potential involvement in the pathogenesis of SLE associated with EBV infection. Notably, IFI27 is considered the most important gene among them. Additionally, our findings suggest that CD4+ T cells, CD8+ T cells, and monocytes may serve as key effector cells playing a vital role in SLE patients infected with the EBV.
REFERENCES: NIL.
Acknowledgements: NIL.
Disclosure of Interests: None declared.