fetching data ... 
Background: Systemic lupus erythematosus (SLE) is a chronic autoimmune disease underscored by complex immune dysregulation, including altered immune mediators and accumulation of autoantibody (AutoAb) specificities. Such immune dysregulation drives the pathogenesis of SLE; patients experience varied disease activity that must be managed to prevent end organ damage that leads to morbidity and early mortality. With the current shortage of rheumatologists and administrative burden of validated clinical disease activity measures, capturing such immune dysregulation associated with clinical disease activity as a lab-based screening test would help prioritize which SLE patients are in need of formal clinical evaluation and follow-up.
Objectives: This study seeks to determine an optimal panel of analytes that distinguishes SLE patients with active disease and refine a Lupus Disease Activity Immune Index (L-DAI).
Methods: We procured samples from patients with classified SLE on dates of low disease activity (<4, range 0-3, n=162; 141 met definition of LLDAS[1]; 21 had prednisone dose >7.5 mg/day) or active disease (≥4, range 4-30, n=162) defined by the hybrid SLEDAI (hSLEDAI)[2]. Race/sex/age-matched healthy control (Ctrl) samples (n=81) were also evaluated. Plasma immune mediators (n=33) were evaluated by microfluidic immunoassay and serum AutoAb specificities, including dsDNA, chromatin, Ro/SSA, La/SSB, Sm, SmRNP, and RNP, were assessed by xMAP assay. The L-DAI is the sum of log-transformed, standardized immune mediators, weighted by the Spearman r correlation coefficient of mediator levels vs. a composite, averaged weighting of hSLEDAI scores and number of AutoAb specificities associated with clinical disease activity. Log-transformed mediator levels were further evaluated using random forest applied machine learning modeling to determine an optimal subset of analytes to inform the L-DAI.
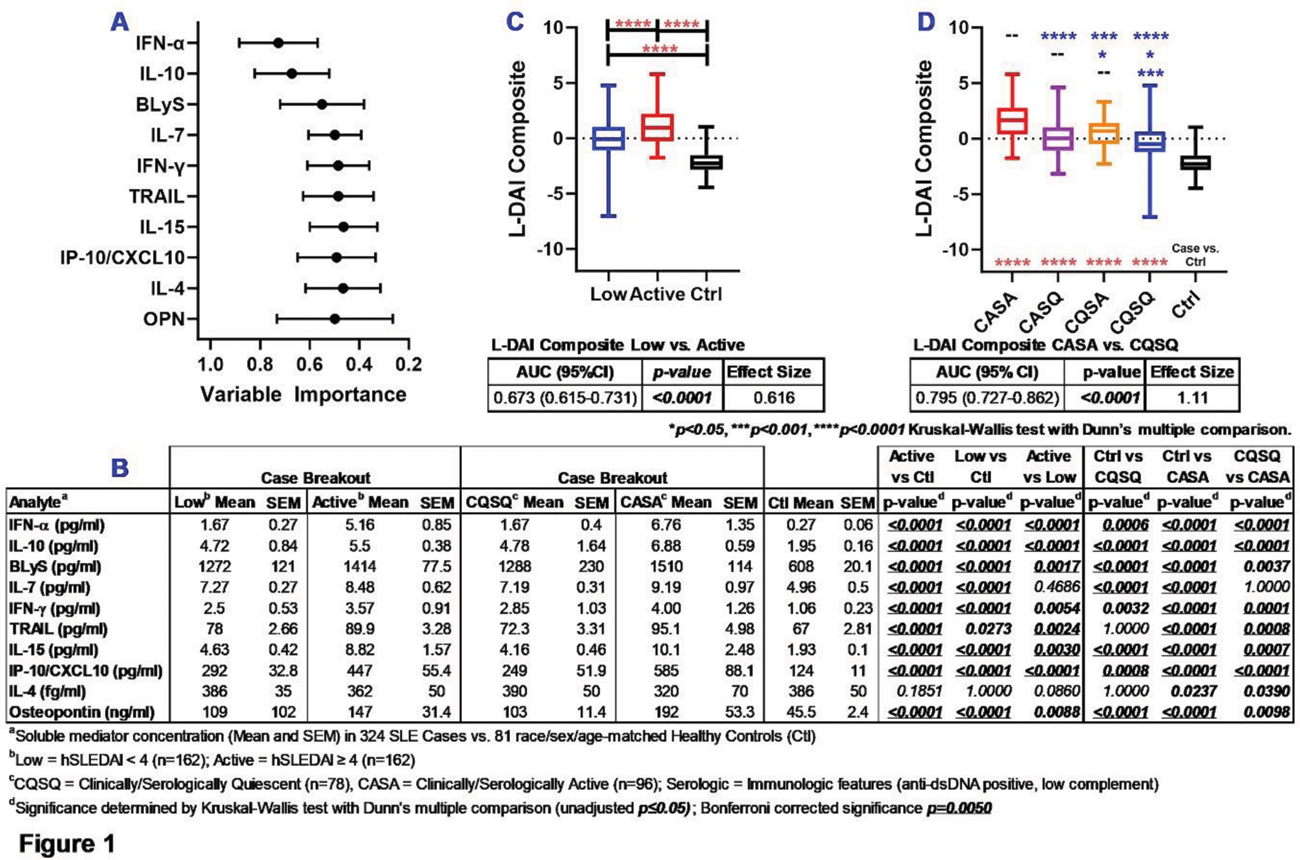
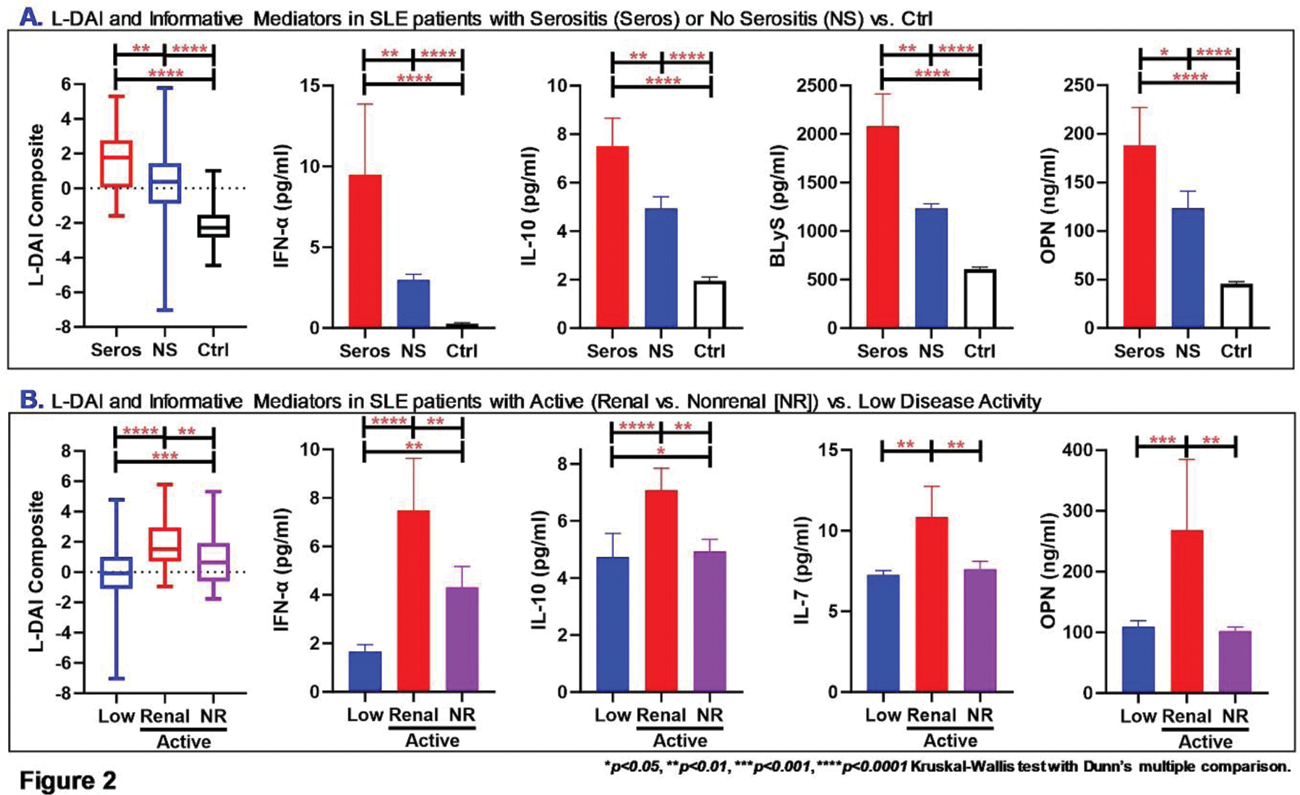
Results: As expected, SLE patients with active disease demonstrated differences in clinical and serologic features, as well as increased use of steroids. Random forest modeling of immune mediators informed a variable importance ranking of 10 mediators that best informed the L-DAI ( Figure 1A , mean ± SD) by differentiating low vs. active disease, including clinically and/or serologically active vs. quiescent disease, and Ctrls ( Figure 1B ). The L-DAI differentiated SLE patients with low, active, and clinically/serologically active vs. quiescent disease (vs. Ctrls, Figure 1C-D ), with moderate (0.616) to large (1.11) effect size and AUC of 0.673 (95% CI 0.615-0.731, p<0.0001 ) differentiating low vs. active disease and 0.795 (0.727-0.862, p<0.0001 ) distinguishing clinically/serologically active vs. quiescent disease. Of interest, the L-DAI differentiated SLE patients with serositis (Seros) vs. non-serositis (NS) features and Ctrls ( Figure 2A ), as well as SLE patients with renal vs. non-renal (NR) active disease vs. low disease activity ( Figure 2B ). IFN-α, IL-10, and Osteopontin (OPN) differentiated SLE patients with both serositis and renal features, while BLyS and IL-7 distinguished serositis and renal features, respectively.
Conclusion: We have refined the L-DAI to characterize SLE patients with active clinical and/or serological disease vs. those with low/quiescent disease. Treat-to-target approaches using a sensitive and objective biomarker surrogate for clinical disease activity has the potential to help improve clinical disease management and prevent organ damage in SLE, as well as characterize SLE patients enrolled in clinical trials and their response to treatment.
REFERENCES: [1] K. Franklyn et al. , Definition and initial validation of a Lupus Low Disease Activity State (LLDAS). Ann Rheum Dis , (2015).
[2] A. Thanou et al. , Impact of heart rate variability, a marker for cardiac health, on lupus disease activity. Arthritis Res Ther 18 , 197 (2016).
Acknowledgements: This study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Institute of Allergy and Infectious Diseases, the National Institute of General Medicinal Sciences, and the National Center for Research Resources of the National Institutes of Health under award numbers M01RR001070 (DLK), P50AR070591 (JPB), R01AR077518 (HZ), R44AI142967 (MEM), P30AR073750 (JAJ), UM1AI144292 (JAJ), and U54GM104938 (JAJ), as well as Oklahoma Center for the Advancement of Science and Technology under award numbers AR16-014 (MEM) and AR18-019 (EJ).The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of Interests: Melissa E Munroe Progentec Diagnostics, Inc. (part time employee), Progentec Diagnostics, Inc., Derek Blankenship Progentec Diagnostics, Inc., Daniele DeFreese Progentec Diagnostics, Inc., Adrian Holloway Progentec Diagnostics, Inc., Mohan Purushothaman Progentec Diagnostics, Inc., Wade DeJager: None declared, Susan Macwana: None declared, Joel M Guthridge: None declared, Stan Kamp: None declared, Nancy Redinger: None declared, Teresa Aberle: None declared, Eliza F Chakravary: None declared, Cristina Arriens AstraZeneca, Aurinia, AstraZeneca, Bristol-Myers Squibb, Cabaletta, GSK, Kezar, UCB, AstraZeneca, Bristol-Myers Squibb, Yanfeng Li: None declared, Hu Zeng: None declared, Stephanie Dezzutti: None declared, Peter Izmirly: None declared, Uma Thanarajasingam: None declared, Diane L. Kamen: None declared, Jill P Buyon Bristol-Myers Squibb, GSK, Related Sciences, Judith A. James GSK, Novartis, Bristol-Myers Squibb, Progentec Diagnostics, Inc., Eldon Jupe Progentec Diagnostics, Inc.