fetching data ... 
Background: Still’s disease (SD) is a complex pathology with autoinflammatory and systemic features and is characterized by the classic triad of fever, arthritis and salmon-pink maculopapular skin rash. It remains a diagnostic and therapeutic challenge due to the absence of definitive criteria. For this reason, new EULAR/PReS 2024 recommendations have been published providing new guidelines to assist the physician in the recognition and follow-up of this disease.
To describe the baseline characteristics of a cohort of patients with SD treated at a tertiary care hospital.
To analyse how many patients started symptoms before the age of 16 years (<16y) and to determine the association with progression to a chronic joint (CJ) course.
To analyse the chronology of patients with SD, assessing previous diagnoses and possible genetic mutation after performing a genetic panel.
Methods: Retrospective descriptive study of 37 patients with SD. We based our study on the new EULAR/PReS 2024 recommendations, unifying Systemic Juvenile Idiopathic Arthritis (SJIA) and adult SD into a single pathology called SD. Yamaguchi criteria (YC) are defined as: major criteria (fever ≥39ºC of ≥ 7 days duration; arthralgia of ≥ 2 weeks duration; typical rash; leukocytosis of ≥ 10,000/mm3, with ≥ 80% granulocytes) and minor criteria (sore throat; lymphadenopathy and/or splenomegaly; liver dysfunction; rheumatoid factor and negative antinuclear antibodies). Five criteria must be met, at least two of the major criteria. Quantitative variables are expressed as mean (std dev) and dichotomous variables as percentages (%). The association of qualitative variables was analysed using the chi-square test.
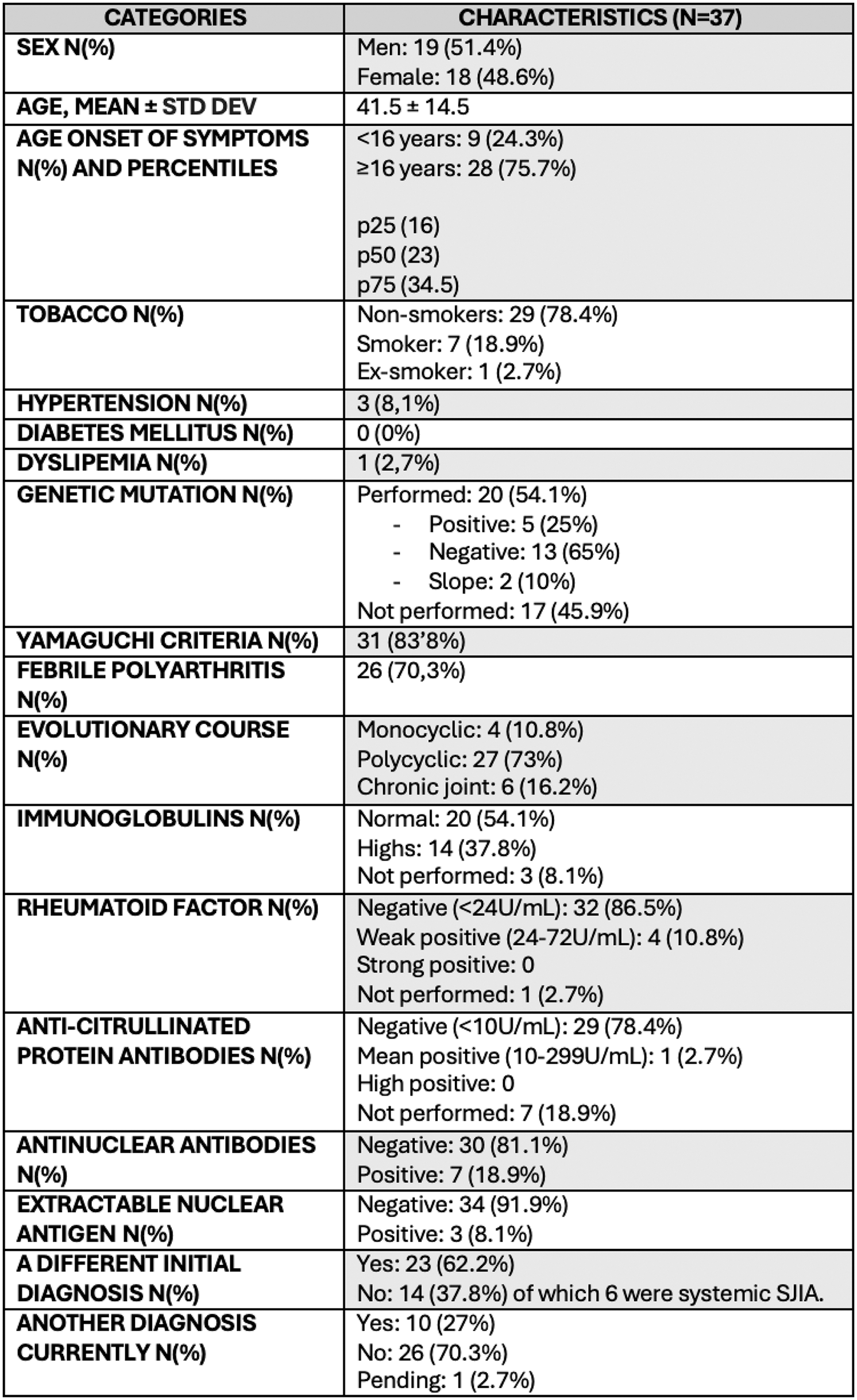
Results: Of the 37 patients with a diagnosis of SD, the mean age was 41.5 ± 14.5 years. Baseline characteristics are described in Table 1. 24.3% of the patients were diagnosed <16y, while 75.7% were diagnosed at the age 16 years or older (≥16y). 83.8% of patients met the YC. 70.3% had febrile polyarthritis. The evolutionary course observed were: 10.8% monocyclic, 73% polycyclic and 16.2% CJ. When analysing the age of symptom onset, 33.3% of patients with CJ started with clinical onset <16y, compared to 10.7% who started later. Although statistical significance was not reached (p=0.3), this finding could be related to the persistence of joint involvement in adulthood in patients with SJIA. In terms of symptoms, 97.3% presented arthralgias and 67.6% arthritis. Other common symptoms were fever (91.9%), odynophagia (48.6%), evanescent salmon-pink rash (78.4%), myalgia (40.5%), abdominal pain (27%), lymphadenopathy (37.8%), splenomegaly (10.8%) and hepatomegaly (8.1%). Among the complications, pleuritis was reported in 5.4%, pericarditis in 8.1%, renal involvement in 2.7%, macrophage activation syndrome (MAS) in 13.5%, and erosive joint involvement in 10.8%; none had interstitial lung disease (ILD). A previous diagnosis other than SD was present in 62.2% and 16.2% started as SJIA. Over time, an autoinflammatory diseases panel was performed in 20 patients (54.1%), of which 5 (25%) showed positive gene mutations. In three cases, the diagnosis of SD was changed to another monogenic autoinflammatory disorder: a TRAPS syndrome (sd), a VEXAS sd and a familial Mediterranean fever (FMF). In total, in 2024, 10 patients (27%) have a diagnosis other than SD.
Conclusion: SD remains a diagnosis of exclusion in clinical practice due to the absence of definitive criteria. The use of YC, widely adopted as a tool for classification, has proven to be useful in identifying the disease. However, its high sensitivity remains a limitation, as reflected by the results of our study, where more than a quarter of the patients initially diagnosed with SD were subsequently reclassified. This phenomenon highlights the importance of considering alternative diagnoses and integrating advanced tests. Future research is needed that focuses on developing and validating specific diagnostic criteria for SD. These criteria should incorporate clinical, analytical and genetic biomarkers to differentiate it from other autoinflammatory and autoimmune diseases with similar manifestations.
Table 1.
REFERENCES: NIL.
Acknowledgements: NIL.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (