fetching data ... 
Background: Rheumatoid arthritis (RA) is the most common chronic inflammatory rheumatic disease, characterized by synovitis associated with progressive bone loss and joint degradation. These few past years, literature showed opposite effects of mechanical stress on RA. We recently observed that mechanical unloading was able to prevent arthritis development in the adjuvant-induced arthritis (AIA) model. The AIA model is widely used for its pathophysiological similarities to RA, its high prevalence and reproducibility. In RA, Yes-associated protein (YAP) is a mechanosensitive transcription factor involved in inflammatory signaling and arthritis progression, especially by driving the inflammatory phenotype of fibroblast-like synoviocyte (FLS). Our previous work showed that CCL2 was also involved in mechanical response. CCL2 is overexpressed in ankles of the AIA model; and also in RA FLS organoids in response to mechanical stress (at a dynamic pressure similar to the intra-articular pressure found in arthritic joint submitted to physical activity).
Objectives: As we already demonstrated that mechanical unloading prevented arthritis development, we hypothesized that the mechanisms involved were dependent from YAP and CCL2. Then, the aim of this study was to determine if blocking YAP and CCL2 was able to recreate the mechanical unloading-induced arthritis prevention in the AIA model.
Methods: Arthritis was induced in 3-4 weeks female rats, defining day (D)0. Arthritis developed on ankles from D10 (onset) to D17 (peak of inflammation). AIA rats were randomised at D6 and received different treatments (n=12/group): a group with verteporfin (VP [20mg/kg IP daily]) from D6 to D12 (VP-D12) or from D6 to D16 (VP-D16), a group with an anti-CCL2 antibody (anti[α]-CCL2 [1 mg/kg intra-articular]) injected at D6, D7, D11, and D15, a group with VP+α-CCL2, and a control group with saline injections. Ankle circumference (mm) and arthritic index (from 0 [normal ankle] to 4 [swollen and disuse ankle]) were used to assess the severity of arthritis. On D17, the ankles were harvested for analysis of bone loss with µCT parameters, and for gene expression of arthritis markers.
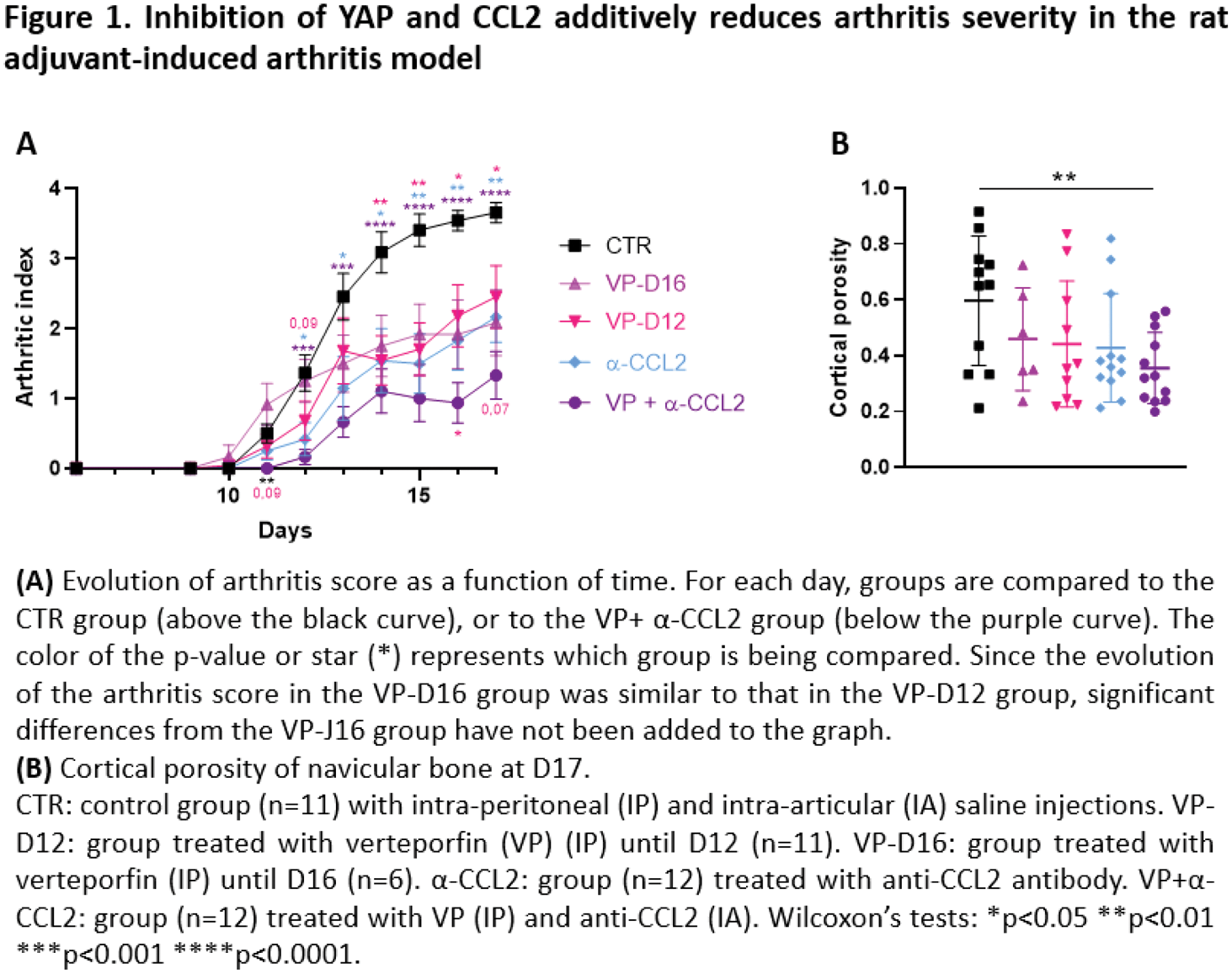
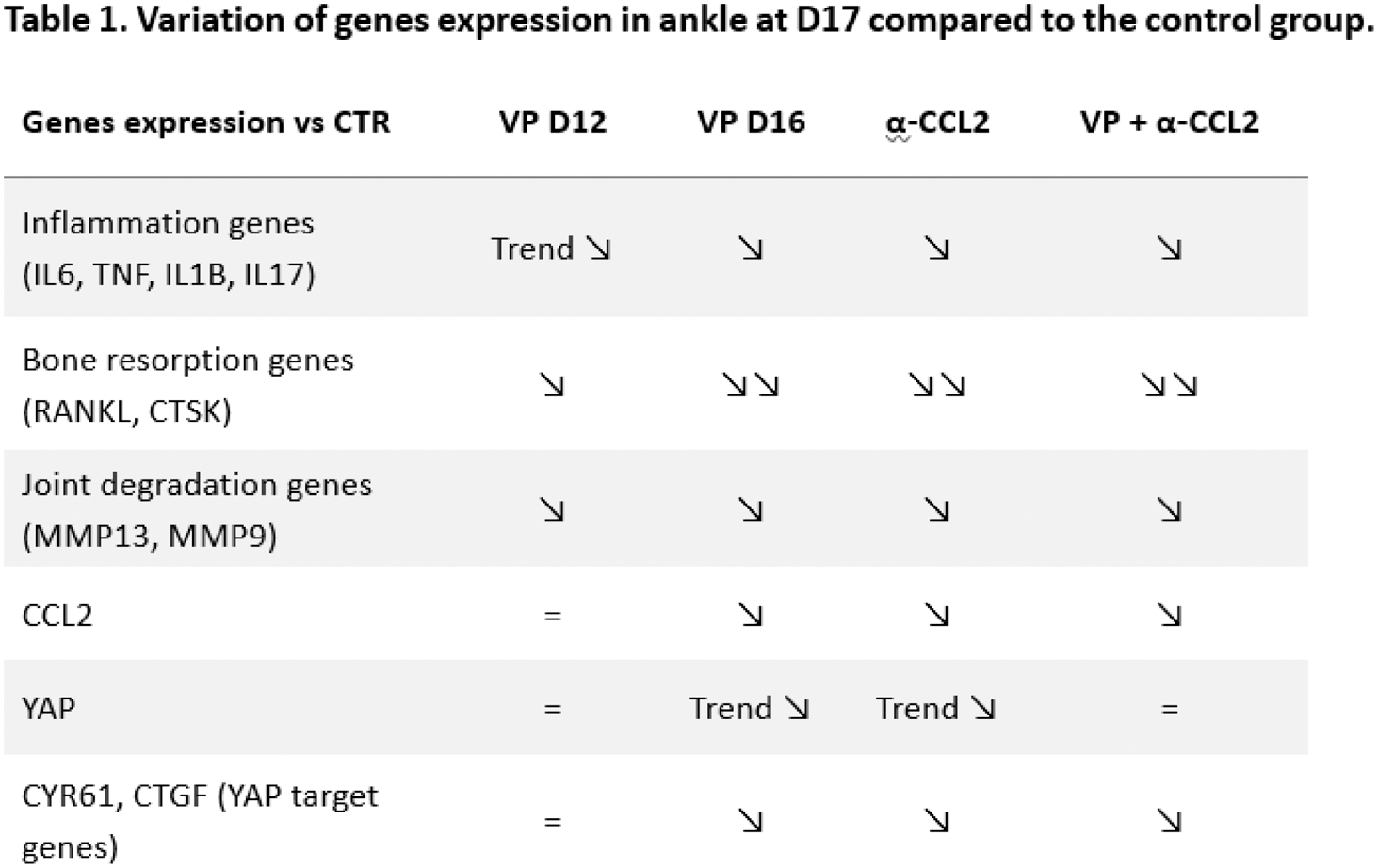
Results: Arthritis started to develop from D10, except in the VP+α-CCL2 group where clinical onset was observed starting from D11 (Figure 1A). At D11, only the VP+α-CCL2 group had an arthritic index significantly lower than the control group (p<0.01). At D12, a trend to decrease arthritic index was observed in the VP group compared to the control group. At D13, ankle circumference was lower in the α-CCL2 group and the VP+α-CCL2 group compared to the control group (p<0.01). VP group, α-CCL2 group, and VP+α-CCL2 group developed a less severe arthritis over time in comparison with the control group (Figure 1). At D17, the control group developed a severe arthritis. In the VP-D12 group, the VP-D16 group and the α-CCL2 group, rats developed a moderate arthritis compared to the control group (p<0.01 and p<0.001, respectively). Arthritis severity over time was not statically different between these 3 groups. However, the VP+α-CCL2 group, rats developed a slight arthritis compared to the control group (p<0.0001), which was significantly decreased at D16 and D17 in the VP+α-CCL2 group compared to the VP-D12 group (p<0.01). Moreover, cortical porosity of navicular bone at D17 was only significantly reduced in the VP+α-CCL2 group compared to the CTR group (p<0.01). In the ankle at D17, expression of inflammatory genes (IL6, TNF, IL1B, IL17) and bone resorption genes (RANKL, CTSK) was slightly reduced in the VP-D12 and greatly reduced in the VP-D16, α-CCL2 and VP+α-CCL2 groups compared to the CTR group (Table 1). Bone degradation genes were similarly reduced in all the treated groups compared to the CTR group. CCL2, YAP and its target genes (CYR61 and CTGF) were similarly expressed in the VP-D12 and the CTR groups, contrary to the VP-D16 and the α-CCL2 groups which had a reduced expression compared to the CTR group. In the VP+α-CCL2 group, only CCL2 and YAP target genes were less expressed compared to the CTR group.
Conclusion: There is a discrepancy between clinical and gene expression assessment in the ankle. The time of VP administration did not alter clinical inflammation or bone loss, whereas VP administration up to D16 decreased the expression of inflammatory genes, bone resorption genes compared with treatment up to D12. YAP inhibition had the same efficiency than CCL2 inhibition on arthritis severity. Inhibition of both YAP and CCL2 had an additive effect on arthritis severity and bone loss decrease over time, which was not found at the level of gene expression in the ankle at D17. We also observed a trend to delay arthritis onset when YAP and CCL2 are both inhibited. However, it did not succeed to prevent arthritis like mechanical unloading, meaning that another mechanism independent from CCL2 and YAP is involved in arthritis development in response to mechanical stress.
REFERENCES: NIL.
Acknowledgements: NIL.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (