fetching data ... 
Background: Changes in serum cholesterol loading capacity (CLC) on macrophages overtime were linked to coronary atherosclerosis progression in rheumatoid arthritis (RA) [1]. Disease-related inflammation, LDL oxidation and autoantibodies were shown to influence macrophage cholesterol loading and coronary plaque burden both directly and indirectly [2]. Oxidized LDL readily formed complexes with beta-2-glycoprotein-1— a protein with antiatherogenic properties readily present in both serum and plaque— in both RA and controls. The complex can be subsequently bound by anti-beta-2-glycoprotein-1 antibodies (anti-b2GPI Ab). We recently reported that anti-b2GPI Ab specifically of the IgA isotype, independently contributed to coronary atherosclerosis progression in RA; yet the precise mechanisms remain speculative [3].
Objectives: We hypothesized that anti-b2GPI IgA may promote atherosclerosis progression by influencing oxidized LDL-mediated cholesterol loading on arterial macrophages.
Methods: Coronary computed tomography angiography evaluated atherosclerosis (noncalcified, mixed or fully calcified plaques) in 100 patients without cardiovascular disease at baseline and follow-up 6.9±0.4 years later. Presence of five or more coronary plaques in a patient and lesions rendering greater than 50% luminal stenosis were considered extensive and obstructive disease respectively. CLC was measured as intracellular cholesterol content in serum treated human THP-1 monocyte-derived macrophages with a fluorimetric assay both at baseline and follow-up assessments. Serum anti-b2GPI IgA were measured with ELISA at baseline visit and positivity was confirmed 12 weeks later. Multivariable negative binomial regression and logistic regression evaluated the influence of anti-b2GPI IgA on the relationship between CLC change and number of new plaques and likelihood of noncalcified plaque formation and new extensive or obstructive disease respectively.
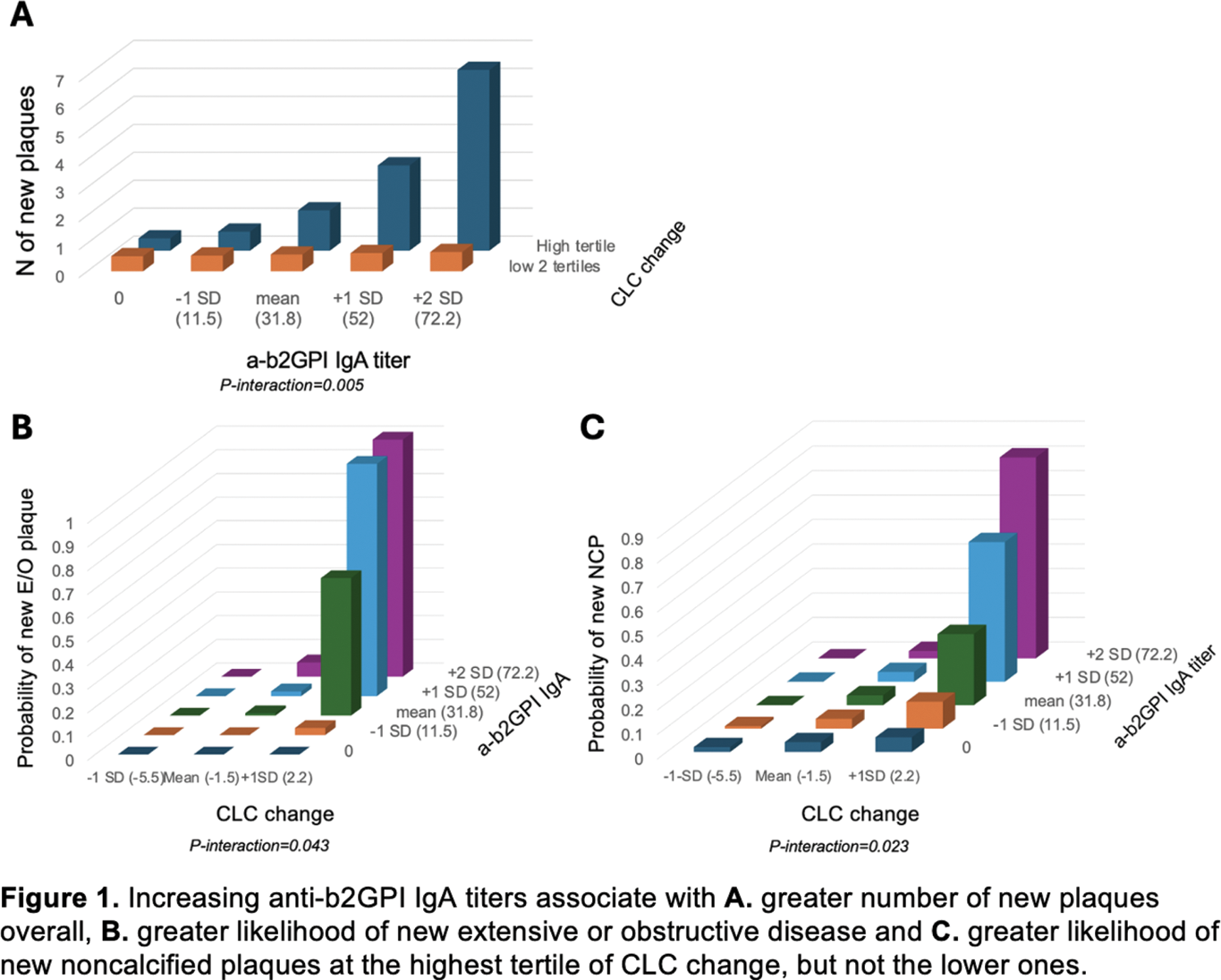
Results: A significant interaction between CLC change and anti-b2GPI IgA titers on plaque progression indicated that the influence of increasing anti-b2GPI IgA titers on atherosclerosis progression varied across levels of CLC change. Specifically, increasing anti-b2GPI IgA titers associated with greater number of new plaques overall (p-for-interaction=0.005), a higher likelihood of new noncalcified plaque formation (p-for-interaction=0.023) and new extensive or obstructive disease (p-for-interaction=0.043) only at the highest tertile of CLC change (Figure 1). This suggested that anti-b2GPI IgA may be actively involved in macrophage cholesterol loading by oxidized LDL in a dose-dependent manner. Presence of anti-b2GPI IgA may also determine the composition of newly formed plaques in the context of increasing CLC change (Figure 2). Specifically, in the presence of anti-b2GPI IgA, CLC change associated with greater likelihood of higher-risk noncalcified plaque progression (odds ratio [OR] 2.82, 95% confidence interval [95%CI] 1.40-5.67, p=0.004 and p-for-interaction=0.005), but not progression of fully-calcified plaque (p=0.364). In contrast, in the absence of anti-b2GPI IgA, CLC change associated with a greater likelihood of lower-risk fully-calcified plaque progression (OR 1.48, 95%CI 1.19-1.84, p<0.001 and p-for-interaction=0.046), but not noncalcified plaque progression (p=0.658)
Conclusion: Anti-b2GPI IgA antibodies may facilitate cholesterol loading onto macrophages and influence coronary atherosclerosis progression and plaque composition in a dose dependent manner, specifically at high levels of CLC change. Our results support a potential mechanism that may involve anti-b2GPI IgA binding of oxidized LDL/b2GP1 complexes to FcA receptors on arterial macrophages, that promotes cholesterol loading and clinically translates to atherosclerosis progression in vivo.
REFERENCES: [1] Karpouzas GA et al. RMD Open. 2024 Dec 24;10(4):e004991
[2] Karpouzas GA et al. Rheumatology (Oxford). 2023 Mar 1;62(3):1254-1263.
[3] Karpouzas GA et al. Semin Arthritis Rheum. 2021 Feb;51(1):20-27.

Acknowledgements: NIL.
Disclosure of Interests: George Karpouzas Scipher, Janssen, Pfizer, Scipher, Bianca Papotti: None declared , Sarah Ormseth: None declared , Marcella Palumbo: None declared , Matthew Budoff: None declared , Nicoletta Ronda: None declared .
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (