fetching data ... 
Background: Rheumatoid arthritis (RA) is a heterogeneous disease in which both environmental and genetic factors contribute to aetiology and disease progression. While genome-wide association studies (GWAS) have successfully identified genetic variants predisposing to RA, with the latest study reporting 124 loci [1], a complete mechanistic understanding of RA remains challenging. The identification of distinct and phenotypically more homogeneous subtypes of RA [2,3] is a significant step forward in our understanding of RA disease heterogeneity and offers new opportunities to improve our functional understanding of variants found through large-scale genetic association studies. Previously, Lewis et al . [2] studied immunohistochemistry assays of synovial tissue of patients from the Pathobiology of Early Arthritis Cohort (PEAC) and identified three subtypes amongst patients with early treatment-naive RA. The resulting pathotypes exhibited different synovial disease states and were accordingly characterized as either lymphoid-rich, myeloid- or macrophage rich (myeloid-rich), and fibroblastic-rich pauci-immune (fibroid-rich). Moreover, synovial transcriptome analyses showed major differences in expression between lymphoid-rich on one hand, and the other two pathotypes on the other, suggesting distinct underlying molecular pathways.
Objectives: We aim to explore how the expression of GWAS-identified RA risk genes relates to phenotypic synovial subtypes of RA.
Methods: RA risk genes are obtained from Ishigaki et al . [1] (GWAS catalog accession ID GCST90132222), comprising 145 SNPs (excluding the MHC region) linked to 181 genes. The GWAS includes 35,871 RA patients and 240,149 control individuals. Paired-end RNA-seq samples from PEAC [2] (87 synovium and 57 blood) are accessed from ArrayExpress under accession code E-MTAB-6141. Transcript abundances are estimated over GENCODE v46/GRCh38 transcripts using Kallisto (version 0.48.0) and tximport (version 1.30.0). After filtering for minimal expression, 130 RA risk genes are expressed in blood and 138 in synovium. Pairwise differential gene expression between pathotypes per tissue type is performed using DESeq2 (version 1.42.0) with sex and pathotype as model covariates and a false discovery rate cut-off at p < 0.05. Gene set enrichment analysis is performed using clusterProfiler (version 4.10.0).
Results: In concordance with earlier findings of Lewis et al ., [2] pairwise differential gene expression analyses showed high numbers of genes upregulated in the lymphoid-rich pathotype compared to either the myeloid- or fibroid-rich pathotype in synovium, but not in peripheral blood (Table 1). A subset of the 138 RA risk genes expressed in synovium is also differentially expressed between lymphoid-rich and either myeloid-rich (N = 51) or fibroid-rich (N = 74) (Table 1), with the majority being higher expressed in the lymphoid-rich pathotype. Differentially expressed RA risk genes show a large overlap (N = 22) and similar effect sizes (Figure 1), which in effect set the lymphoid-rich apart from the other two pathotypes. Compared to other differentially expressed genes, RA risk genes were more likely to be significantly upregulated in both lymphoid-rich vs. myeloid-rich (p adj = 5.02 x 10-3) and fibroid-rich (p adj = 2.53 x 10-4).
Number of differentially expressed genes in blood and synovium.
| Synovium | All genes
| RA risk genes (N = 130) | Blood | All genes (N = 18777) | RA risk genes (N = 138) |
|---|---|---|---|---|---|
| Lymphoid-rich (+) vs myeloid-rich (-) | + 2512
| + 44
| + 22
| + 0
|
|
| Lymphoid-rich (+) vs fibroid-rich (-) | + 4612
| + 59
| + 0
| + 0
|
|
| Fibroid-rich (+) vs myeloid-rich (-) | + 176
| + 0
| + 135
| + 7
|
Correlation between LFC of differentially expressed RA risk genes in synovial tissue between pathotypes. LFC = log fold change.
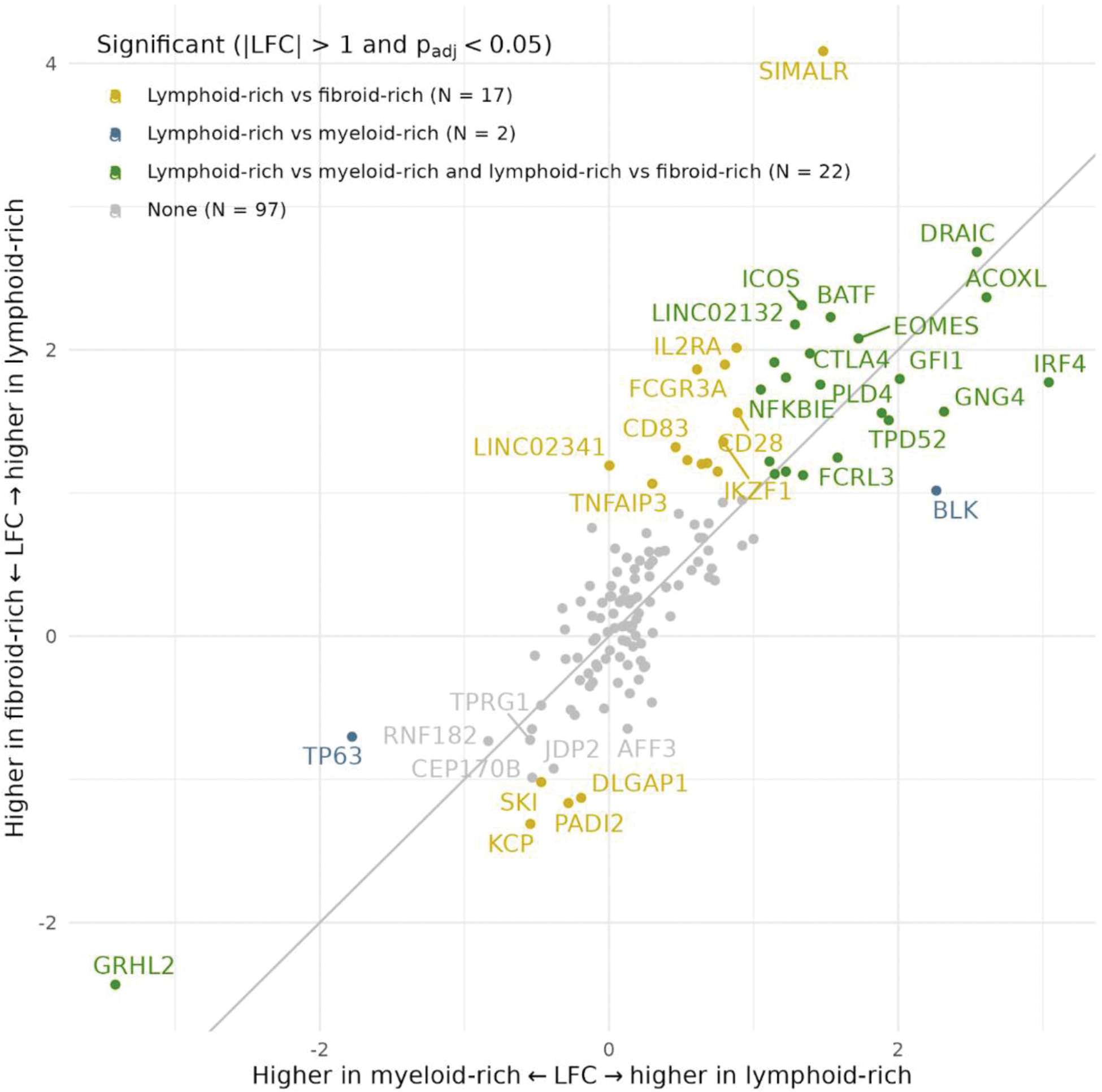
Conclusion: We show that GWAS-identified RA risk genes are enriched in the lymphoid-rich pathotype compared to the myeloid-rich and fibroid-rich pathotype, potentially giving insights into underlying molecular mechanisms of these disease states. Consistently upregulated RA risk genes included those with well-described roles in lymphocyte differentiation, including ICOS, BATF and IRF4, but also included the long non-coding RNA DRAIC. Other work on the cohort has shown that several significant eQTL in synovial tissue have previously been identified by GWAS to be associated with RA, osteoarthritis, and autoimmune diseases [4]. Interestingly, these reported genes are different from the RA risk genes that we find significantly differentially expressed between lymphoid-rich and myeloid- or fibroid-rich synovial tissue. Our findings could point to the preferential inclusion of the lymphoid-rich pathotype in case-control designed GWAS studies, which would result in a larger power to discover predispositions for the lymphoid-rich pathotype specifically. Collectively, our findings highlight the need for RA patient subtyping in the design of genetic and functional studies of RA.
REFERENCES: [1] Ishigaki, K. et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 54, 1640–1651 (2022).
[2] Lewis, M. J. et al. Molecular Portraits of Early Rheumatoid Arthritis Identify Clinical and Treatment Response Phenotypes. Cell Rep. 28, 2455-2470.e5 (2019).
[3] Zhang, F. et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature 623, 616–624 (2023).
[4] Goldmann, K. et al. Expression quantitative trait loci analysis in rheumatoid arthritis identifies tissue specific variants associated with severity and outcome. Ann. Rheum. Dis. 83, 288–299 (2024).
Acknowledgements: This project has received funding from Horizon Europe programme under grant agreement no. 101080711 (SPIDeRR), ZonMW klinische fellow no. 40-00703-97-19069, and ZonMw Open Competitie 2021 no. 09120012110075.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (