fetching data ... 
Background: The pathogenesis of rheumatoid arthritis (RA) is predominantly attributed to an autoimmune reaction against citrullinated proteins, coupled with aberrations in T-cell differentiation, which are influenced by a confluence of genetic predispositions and environmental factors.
Objectives: To identify increased immune cell subsets in RA patients via comprehensive immune phenotyping, covering T cells, B cells, NK cells, dendritic cells, and monocyte subsets, and to investigate the implications of these cellular anomalies.
Methods: We conducted immunophenotyping of RA patients’ blood using multi-color flow cytometry and investigated factors contributing to the abnormal cell differentiation in vitro.
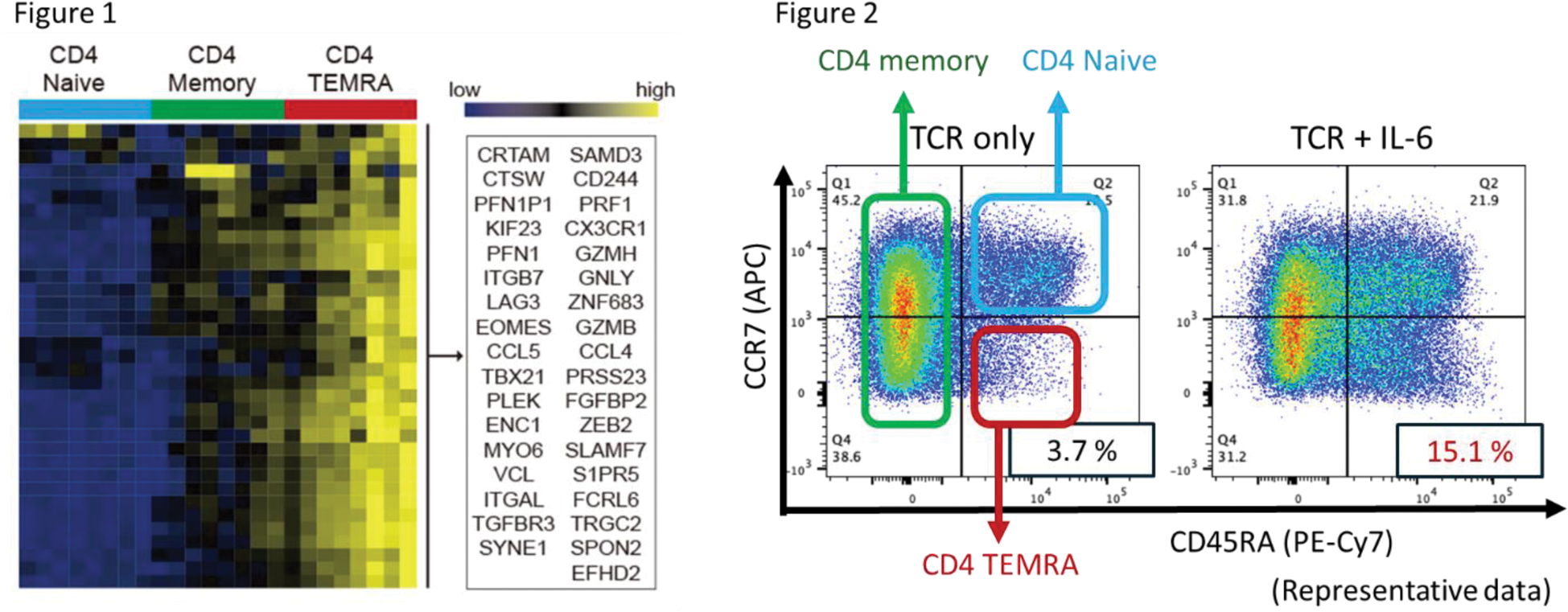
Results: In a comparative study involving 533 patients diagnosed with RA who had never used b/tsDMARDs and 96 healthy individuals, we observed alterations primarily centered around abnormal T-cell differentiation in the patients with RA, with a marked increase in CD4 + effector memory T cells re-expressing CD45RA (CD4 + TEMRA) cells. To explore the role of these cells in pathogenesis, CD4 + TEMRA (CD4 + CCR7 - CD45RA + ) cells were isolated from peripheral blood for transcriptome analysis. These analyses revealed that CD4 + TEMRA cells exhibited a high expression of cytotoxic molecules and displayed cytotoxic CD4 + cytotoxic T lymphocytes (CD4 + CTL)-like functions (Figure 1). Additionally, they prominently expressed p16, a marker indicative of cellular senescence. Furthermore, analysis of the T-cell receptor (TCR) repertoire of these cells revealed a reduced diversity in the TCRs of CD4 + TEMRA cells compared to both CD4 + naïve T cells and CD4 + memory T cells, across both the α and β chains. This suggests an oligoclonal expansion of CD4 + TEMRA cells. It has been demonstrated that the inclusion of hydrophobic amino acids (such as tryptophan, phenylalanine, and leucine) in the CDR3 region of TCRs in T cells may facilitate the recognition of self-antigens. Consequently, we analyzed the amino acid sequences constituting the TCRs and discovered a frequent utilization of hydrophobic amino acids in the TCRs of CD4 + TEMRA cells. TCR of regulatory T cells (Tregs) are known for self-reactivity, and when compared to the TCRs of Tregs, CD4 + TEMRA cells demonstrated a high degree of homology with these TCRs. However, subsequent analysis using Foxp3 and CD25 staining to detect the percentage of Tregs revealed that CD4 + TEMRA cells contained almost no Tregs. In other words, it was suggested that the CD4 + TEMRA population exhibits oligoclonal expansion and autoreactivity. In vitro experiments, when naïve T cells were stimulated through TCR activation, some were induced to differentiate into CD4 + TEMRA cells, expressing both cell senescence markers (p16 and CD57) and cytotoxic molecules. When the telomere length of these CD57-positive cells was detected by PCR, a shortening was observed, confirming that CD4 + TEMRA were senescent cells. When IL-6 was added to TCR stimulation, the differentiation into CD4+ TEMRA was notably accelerated, increasing from 3.7% to 15.1% (Figure 2). This enhancement was reversed by the administration of tocilizumab and JAK inhibitors. Conversely, IFNγ, IL-12, and IL-17 had no impact on this differentiation. Finally, we investigated the expression of p16 in patients with RA following the administration of molecular targeted therapies. Notably, the expression of p16 in T cells was reduced by the IL-6 inhibitor.
Conclusion: Our study identified an elevation of CD4 + TEMRA cells in RA patients and highlighted the pivotal role of IL-6 in inducing cellular senescence in T cells and promoting their differentiation into autoreactive phenotypes. These findings emphasize the potential therapeutic benefits of targeting IL-6 to mitigate its effects on T cell aging and differentiation, thereby addressing a critical pathway in RA pathogenesis.
REFERENCES: NIL.
Acknowledgements: NIL.
Disclosure of Interests: Satoshi Kubo has received speaking fees from Eli Lilly, Bristol-Myers, GlaxoSmithKline, Abbvie, and also research grants from Daiichi-Sankyo, Abbvie, Boehringer Ingelheim, and Astellas, Katsuhide Kusaka: None declared, YUYA FUJITA: None declared, Nishino Takahiro: None declared, Yusuke Miyazaki has received consulting fees from Astra-Zeneca, speaking fees, and/or honoraria from Eli Lilly and GlaxoSmithKline, Kazuyoshi Ishigaki: None declared, Michihiro Kono: None declared, Hiroaki Tanaka: None declared, Hidenori Sakai: None declared, Masanobu Ueno: None declared, Yasuyuki Todoroki: None declared, Yurie Satoh-Kanda: None declared, Ippei Miyagawa: None declared, Shingo Nakayamada has received speaking fees from Bristol-Myers, UCB, Astellas, Abbvie, Eisai, Pfizer, and Takeda, and also research grants from Mitsubishi-Tanabe, Novartis, and MSD, Yoshiya Tanaka has received consulting fees, speaking fees, and/or honoraria from Abbvie, Daiichi-Sankyo, Chugai, Takeda, Mitsubishi-Tanabe, Bristol-Myers, Astellas, Eisai, Janssen, Pfizer, Asahi-Kasei, Eli Lilly, GlaxoSmithKline, UCB, Teijin, MSD, and Santen, and also research grants from Mitsubishi-Tanabe, Takeda, Chugai, Astellas, Eisai, Taisho-Toyama, Kyowa-Kirin, Abbvie, and Bristol-Myers. Hiroaki Tanaka, Yuya Fujita, Hidenori Sakai, Masanobu Ueno, Yasuyuki Todoroki, Yurie Satoh-Kanda, and Ippei Miyagawa have nothing to declare.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (