fetching data ... 
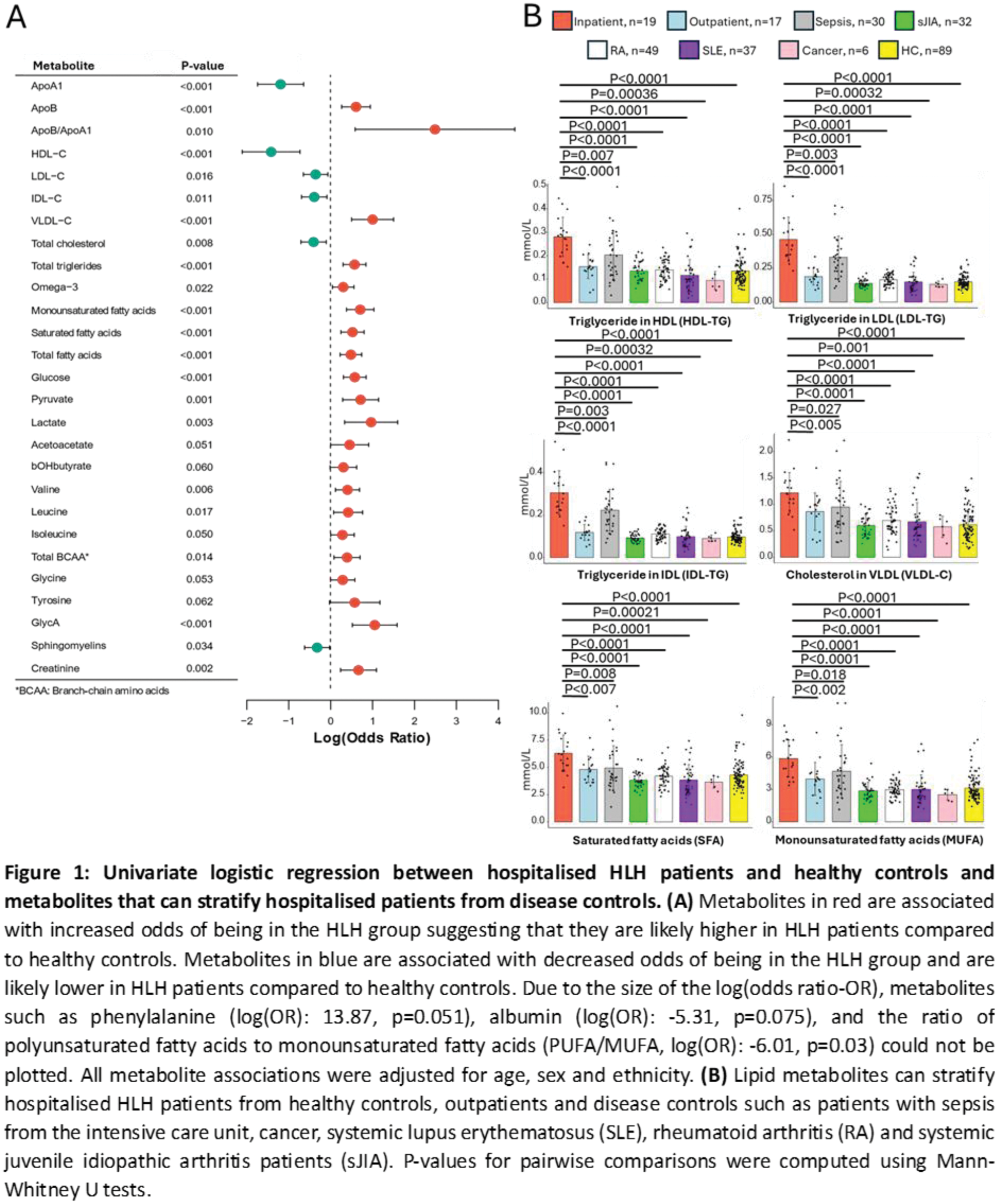
Background: Haemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome with genetic and acquired forms, characterised by cytotoxic lymphocyte defects and uncontrolled activation/proliferation of CD8+ T cells and macrophages. In HLH, hypertriglyceridemia and dampened high- and low-density lipoprotein cholesterol (HDL-C/LDL-C) levels have been reported, suggesting lipid metabolism disturbances. Despite this, a comprehensive study into HLH patient metabolism is lacking.
Objectives: This study used metabolomics and whole blood transcriptomics from matched patients to characterise dysregulated metabolic pathways, potentially associated with secondary HLH (sHLH) pathogenesis.
Methods: Serum metabolites (n=250) from 36 patients with sHLH and 89 age/sex/ethnicity-matched healthy controls (HCs) were quantified using NMR-spectroscopy. Patients were stratified as hospitalised (inpatient) and recovered (outpatient), with both groups on immunomodulatory therapies. Metabolomic data was analysed using supervised machine learning (ML), univariate logistic regression to stratify patients and metabolite enrichment analysis. Results were validated by applying metabolic module analysis on differentially expressed genes (DEGs) between inpatients (n=21), outpatients (n=18) and HCs (n=13). Lastly, gene-set enrichment analysis (GSEA) was conducted and metabolomic and transcriptomic results were integrated using interaction networks.
Results: Patients hospitalised with sHLH were stratified from HCs with high accuracy (91.67-100%) using ML. Hospitalised patients had significantly elevated creatinine, apolipoprotein-(Apo)B:ApoA1, triglycerides (TG), very-(V)LDL-C, LDL-TG, intermediate-(I)DL-TG, fatty acids, branch chain/aromatic amino acids, glycolysis-related metabolites and glycoprotein acetyls (glycA, inflammatory marker) and reduced albumin, sphingomyelins, HDL-C and LDL-C, compared to HCs (Figure 1A). Recovered patients also maintained significantly higher glycA, LDL-TG, IDL-TG, ApoB:ApoA1, creatinine and lactate levels than HCs. Comparing inpatients with recovered patients, 93 metabolites were significantly different after adjusting for age, sex, ethnicity and treatment including ApoB/ApoA1 (odds ratio (OR)=53.62, p=0.019); glucose (OR: 3.58, p=0.014); glycA (OR: 3.07, p=0.031), and many lipid metabolites could stratify inpatients from HLH-mimics (sepsis) and HLH-drivers (e.g. rheumatic diseases) (Figure 1B), suggesting that HLH patients have a unique metabolic signature. Metabolic module analysis identified 1859 DEGs associated with 36 up- and 4 downregulated metabolic pathways in hospitalised versus recovered patients, including glycosphingolipid (p=1.06E-03), triacylglycerol (p=1.09E-04) and fatty acid biosynthesis (p=2.46E-04); tyrosine (p=1.48E-05) and leucine (p=3.89E-05) degradation; glycolysis (p=2.05E-03) and gluconeogenesis (p=1.85E-03) which recapitulated metabolite enrichment analysis results (Figure 2A). The importance of metabolic signalling was reinforced by GSEA which identified heme metabolism (padj=5.40E-08); mammalian target of rapamycin complex 1 (mTORC1) signalling (padj=6.90E-13); G2/M checkpoints (padj=1.60E-18), which are regulated by mTORC1 and E2F targets (padj=2.20E-23) whereby E2F1 (log2foldchange: 1.91, padj=6.51E-09) is a mTORC1 transcription factor. Lastly, gene-metabolite networks identified crosstalk between 60 up- and 8 downregulated genes, (including mTORC1 signalling genes) with 15 metabolites (Figure 2B), which were integrated into glycolysis, gluconeogenesis, cholesterol, fatty acid, lipid, amino acid, iron and glutathione metabolism pathways (Figure 2B).
Conclusion: HLH patients have disrupted metabolic pathways irrespective of disease severity/treatment. These results suggest that immune cells perpetuate hyperinflammatory responses in hospitalised patients by activating mTORC1 through extracellular amino acid acquisition via transporters. As a result, mTORC1 can induce lipid biosynthesis and glycolysis, needed for inflammasome activation and proinflammatory cytokine expression.
REFERENCES: NIL.

Acknowledgements: NIL.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (