fetching data ... 
Background: Chimeric antigen receptor (CAR)-T cells have revolutionized the treatment of hematological cancers and recently emerged as a potential cure for systemic autoimmune diseases [1] but their widespread application in rheumatology is limited by costly manufacturing, lymphodepletion and poor control over biodistribution and pharmacodynamics. As the immune reset in autoimmunity does not appear to require long-term CAR-T engraftment, mRNA technology bears the promise for transient CAR-T cell treatments with an improved safety and controllability over long-term engrafting CAR-T cells [2]. Because in-vitro-transcribed mRNA bypasses the nuclear pathway, it also allows for safe and efficient co-expression of multiple proteins without DNA double strand breaks or any risk of insertional mutagenesis [3]. This opens unique opportunities for multifunctional cell products with higher treatment precision and advanced mode of action tailored to autoimmune indications.
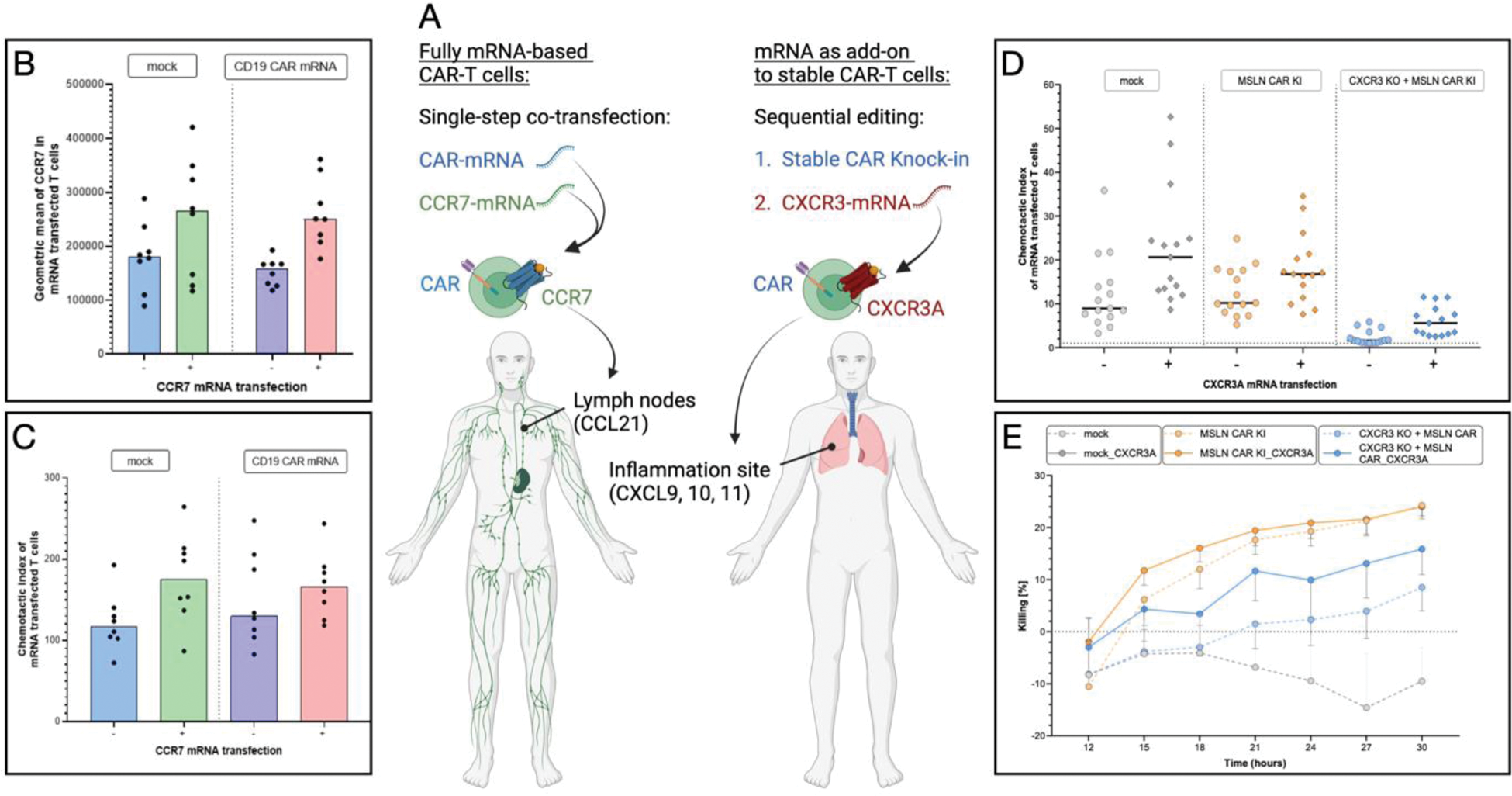
Objectives: We aim to develop next-generation CAR-T cell therapeutics tailor-made for rheumatology, based on non-viral, non-integrating mRNA technology. These cell products are designed to possess improved treatment controllability and safety and targeted migration for deeper B cell depletion in lymph nodes and inflamed tissues (Figure 1A). We optimize mRNA delivery to primary human T cells using lipid nanoparticles (LNPs) versus electroporation. We develop CAR-T cells co-expressing mRNA-based CD19-CAR or stable CRISPR-Cas9 CAR knock-in (KI) with mRNA-encoded chemokine receptors for deeper B cell depletion in lymphatic tissues (CCR7) and transient targeting of inflammation [4]. Finally, we aim to characterize those cell products and compare their functionality to conventional CAR-T cells.
Methods: mRNA was transcribed in vitro [3], LNPs were formulated using microfluidic mixing. Primary human T cells were activated and transfected by LNPs vs electroporation with CAR-mRNA and mRNA encoding CCR7 or CXCR3A. For stable CAR-T cells, CAR was knocked into the T cell receptor alpha chain locus using CRISPR-Cas9 and CXCR3 mRNA was transfected 6 days later. Transfection efficacy and cell viability, mRNA expression kinetics (flow cytometry) and changes in cytokine production (ELISA) were quantified. CAR-T cell functions were compared between migration-engineered CAR-T cells, CAR-only-transfected cells and untreated T cells: CAR-mediated killing of B cells, CAR-T cell migration indices toward target cytokines (CXCL11 for CXCR3A; CCL21 for CCR7), and a combination of migration toward target cells and target elimination were quantified in cell-based human in vitro assays.
Results: mRNA-LNPs were homogenous, stable and functional at 4°C for at least 7 days. LNP-mediated mRNA transfection resulted in effective transfection (>80%) and high viability (>90%) of primary human T cells, performing on par with gold-standard electroporation. In conditions with stable CAR expression, CRISPR-Cas9 CAR-KI rate was >20% and CAR-T cells could be re-transfected with mRNA 6 days post-KI without significant viability loss. In “mRNA-only” CAR-T cells, co-expression of CAR and chemokine receptor mRNAs could be achieved in 80% of cells upon single treatment. The secretory phenotype including IL-2, IFN-g or TNFa was not affected by mRNA transfection. T cells co-transfected with CAR-mRNA and chemokine receptor-mRNA expressed both proteins for ~3d and thereafter assumed their original phenotype. Transfection with CCR7-mRNA strongly boosted the migration of (CAR-)T cells toward the CCL21 chemokine, a marker of secondary lymphoid organs (Figure 1C), while CXCR3A-mRNA increased migration toward the inflammatory mediator CXCL11 (Figure 1D). Early differentiated T cell subsets exhibited enhanced migration toward both chemokines. Chemokine-engineered CAR-T cells outperformed T cells transfected with CAR only in a combined killing and migration assay (Figure 1E), demonstrating a more targeted mode of action of multiplexed CAR-T cells compared to conventional CAR-T cells.
Conclusion: We demonstrate successful engineering of mRNA-based CAR-T cells with a fully transient phenotype and no transfection-related toxicity, as a key safety features. These CAR-T cells could remain active for several days, and their potential to eliminate tissue-resident B cells may be enhanced by the co-expression of tissue-targeting chemokine receptors. Such products differ significantly from conventional CAR-T cells used in oncology and promise improved controllability and safety through re-dosing and a mode of action tailored to deep B cell depletion in autoimmunity. Validation in relevant disease models and transition to GMP-compatible manufacturing are ongoing. Open questions include the optimal re-dosing regimen for transient CAR-T cells and the possibility for in vivo CAR-T cell production using off-the-shelf LNP infusion.
REFERENCES: [1] Mackensen, A, Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med, 2022.
[2] Baker, D.J.,CAR T therapy beyond cancer: the evolution of a living drug. Nature, 2023.
[3] Drzeniek, N.M In Vitro Transcribed mRNA Immunogenicity Induces Chemokine-Mediated Lymphocyte Recruitment and Can Be Gradually Tailored by Uridine Modification. Adv Sci (Weinh), 2024.
[4] Vollmer, T. The intratumoral CXCR3 chemokine system is predictive of chemotherapy response in human bladder cancer. Sci Transl Med, 2021.
Acknowledgements: NIL.
Disclosure of Interests: Samira Picht: None declared, Lisa Hemmerling: None declared, Antonia Maria Klaas: None declared, David Simon AbbVie, Alfasigma, Bristol-Myers Squibb, Janssen-Cilag, Lilly, Novartis, UCB, Arnd Kleyer: None declared, Manfred Gossen: None declared, Hans-Dieter Volk TC Balance, Gerhard Krönke: None declared, Michael Schmueck-Henneresse: None declared, Norman Michael Drzeniek: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (