fetching data ... 
Background: Despite advances in the management of rheumatoid arthritis (RA) and the plethora of targeted therapies now available, up to 21% of patients remain refractory to treatment. This lack of efficacy presents a major clinical challenge, as alternative strategies for managing uncontrolled disease are limited. A wide range of evidence suggest that RA is a T-cell driven disease with CD4+ T-cells playing a central role; however, a comprehensive understanding of their regulation in refractory RA (rRA) is lacking.
Objectives: Using single-cell multiomic analysis, this study aims to delineate the cellular basis and regulation of CD4+ T-cells between treatment-naïve early RA (eRA) and patients with refractory disease. By mapping the molecular features and uncovering new mechanisms underlying treatment resistance, these findings aim to provide a foundation for the development of targeted and individualised treatment strategies.
Methods: Primary CD4+ T-cells were isolated from PBMCs obtained from rRA patients (defined as per EULAR definition of difficult-to-treat RA) and eRA patients, all seropositive with active disease (DAS28-ESR>3.2), collected as part of the PRECISE-RA programme. Single nuclei were captured and processed using 10x Genomics Multiome assay to simultaneously determine the transcriptional (RNA-seq) and chromatin landscapes (ATAC-seq) in individual cells. Data was analysed using Seurat and Signac R packages.
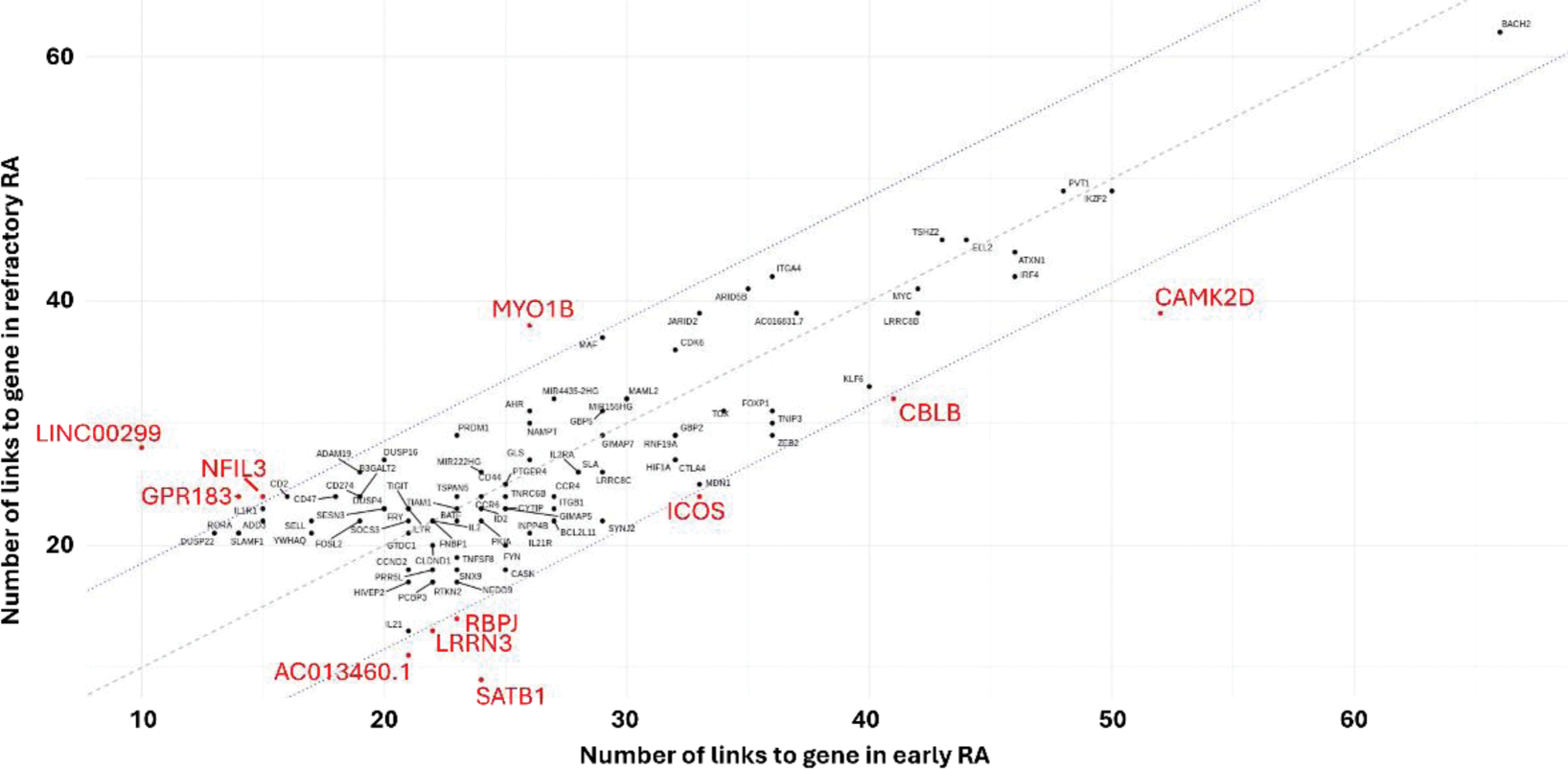
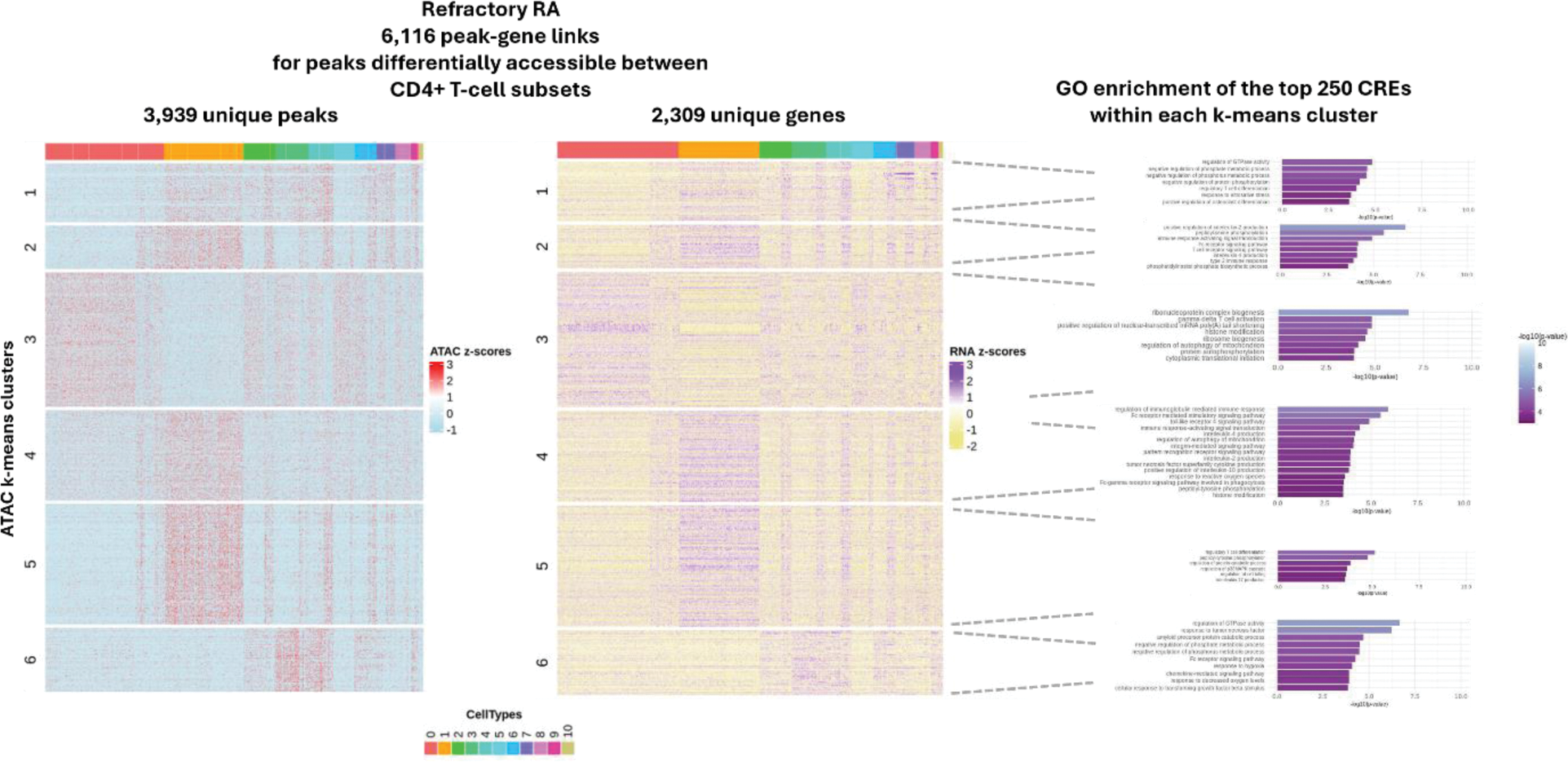
Results: 7,997 cells from 3 rRA and 3 eRA patients were used for this analysis with clinical data shown in Table 1. Using a weighted combination of RNA and ATAC, 12 known cell states were observed which broadly split into naïve, memory, active, resting, Th2 and Tregs. Cis-regulatory elements (CREs) were identified by linking ATAC peaks to target genes (peak-to-gene links), revealing relationships between regulatory elements (e.g. enhancers) and their associated gene expression. We identified a subset of highly regulated genes (HRGs), likely containing critical regulatory sequences, linked to numerous peaks. Among these, BACH2, a transcription factor (TF) essential for CD4+ T-cell differentiation/function, emerged as the most regulated HRG within the dataset (67 links). Other notable TFs included IKZF2, ARID5B, KLF6, MAF, and ZEB2. Comparison of HRGs between eRA and rRA, identified 11 genes as differentially regulated (Figure 1). These included CAMK2D, CBLB, ICOS, RBPJ and SAT1B, which play roles in chromatin organisation, activation and regulation of signalling. A notable less regulated gene included CCR2 which mediates cell migration (12 and 0 links in eRA and rRA, respectively). Next, we explored regulation on a more global scale by clustering ATAC-seq peaks from all CREs to identify co-accessible regulatory modules and gene ontology (GO) on the linked genes to biologically characterise the modules. We observed regulatory modules to be shared across cells with varying enrichment across different T-cell states. In rRA we found enrichment for 2 modules which were not observed in early RA (Figure 2). One, associated with negative regulation of protein phosphorylation, phosphate and phosphorus metabolic processes and Treg differentiation; indicating heightened activation and efforts to modulate excessive immune responses. The other, enriched in processes such as ribosome biogenesis, cytoplasmic translational initiation and histone modification; which likely reflect the increased need in proteins/RNA due to heightened activation. We then focused on GO terms unique to eRA or rRA. Notably, unique eRA terms mainly clustered within a single regulatory module, while no such enrichment was observed in rRA. This suggests more organised regulation in eRA, whereas rRA appears more heterogeneous, with less centralisation, indicating that treatment strategies may need to be broader. Finally, we sought to identify differentially active TFs between eRA and rRA by correlating motif activity with TF gene expression and identify potential target genes. Of the 6 TFs identified, BACH1, CREM, FOSL1, RUNX2, SMAD2 and STAT1, all displayed a stronger correlation in rRA. LGALS3 (Galectin-3) emerged as a target gene for BACH1, FOSL1 and RUNX2, but only in rRA. Previous studies have linked serum LGALS3 to HAQ scores, with lower concentrations correlating with better physical function; Galectin-3 inhibitors are currently in clinical trials for fibrotic diseases and psoriasis.
Conclusion: These results, to be validated in a larger cohort, suggest that CD4+ T-cells in rRA are undergoing significant molecular changes which could sustain or exacerbate the autoimmune and inflammatory processes and has the potential to elucidate novel avenues for further exploration into RA pathogenesis, biomarker discovery and therapeutic opportunities.
REFERENCES: NIL.
Comparison of highly regulated genes (HRGs) in early RA (x-axis) and refractory RA (y-axis).
Co-accessible regulatory modules observed in refractory RA.
Acknowledgements: This research was funded by the NIHR Manchester Biomedical Research Centre (NIHR203308). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. This work was also supported by the Centre for Genetics and Genomics Versus Arthritis (UK grant number 21754) at the University of Manchester and has received support from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 101007757 (HIPPOCRATES).
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (