fetching data ... 
Background: Common variable immunodeficiency (CVID) is the most frequent clinically symptomatic primary immunodeficiency. Its clinical spectrum is highly variable, ranging from isolated recurrent infections to autoimmune (AID) and inflammatory rheumatic diseases (IRD), which may even be the unique manifestation at disease onset [1]. CVID is mainly a polygenic disease, even if recent studies employing whole-genome and exome sequencing have highlighted that 15–30% may display a monogenic origin [2], even if not all these mutations have been validated from a functional point of view.
Objectives: The aim of the study is to describe our cohort of CVID patients (pts), highlighting AID, IRD and related immunosuppressive (IS) treatment; we also aimed at describing genetic variants possibly linked to the condition of immunodeficiency and autoimmunity and their possible therapeutic implications.
Methods: This is an observational retrospective study considering CVID pts followed in a single centre since 1985 to 2024. Diagnosis was made according to European Society for Immunodeficiency criteria [3]. Demographic, clinical, in particular concomitant AID diagnosis including IRD, radiologic, laboratory and histologic data were retrieved from clinical charts. A next generation sequencing (NGS) analysis of genes potentially linked to hypo-agammaglobulinemia and to antibody deficiency was executed when considered clinically appropriate and with pts’ informed consent. Continuous variables are presented as median (interquartile range) and were compared using non-parametric tests (Mann-Whitney or Wilcoxon test). Categorical variables are presented as number (percentages) and were compared using Chi-Square or Fisher exact test.
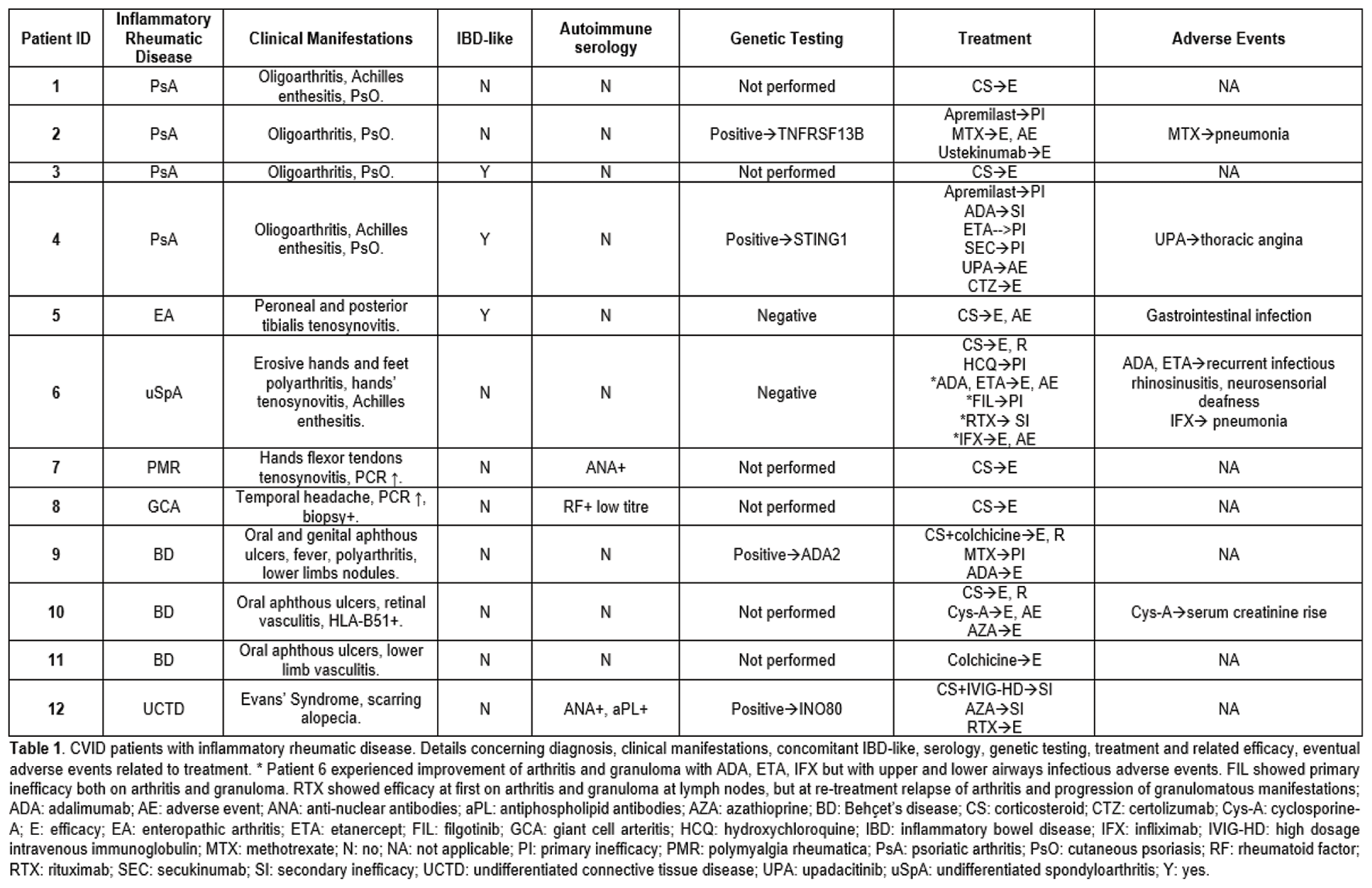
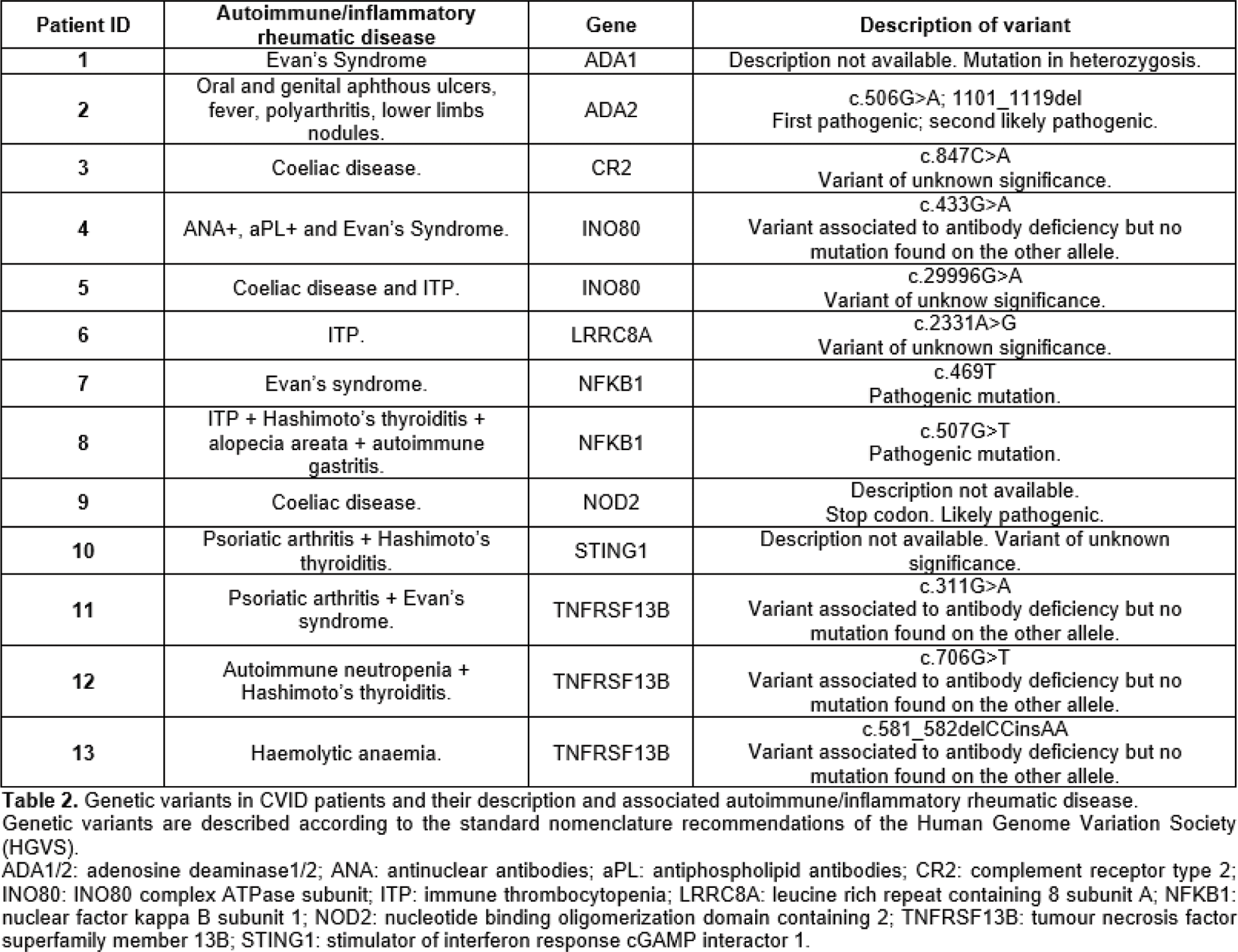
Results: Eighty-three pts with a CVID diagnosis were considered; female to male ratio was 1.2 (54.2% vs 45.8%). All patients were Caucasian; 4.8% had a familiarity for immunodeficiency, being in all cases selective IgA deficiency. Thirteen among 83 (15.7%) also had a familiarity for AID, organ-specific and/or systemic. Median age at disease onset was 33.0 (21.5-41.5) years and median age at diagnosis was 39.0 (29.0-46.0) years. Among the whole cohort, 9.6% of the pts had died. AID, including both organ-specific and/or systemic, had a prevalence of 55.4% in the whole cohort. When comparing CVID pts with AID and without, the formers were younger [49.0 (42.3-55.0) vs 59.0 (47.0-65.0); p:0.031], with lower age at disease onset [25.0 (16.8-40.0) vs 38.0 (29.0-43.0); p:0.025] and at diagnosis [35.0 (24.5-45.0) vs 43.0 (34.0-46.0); p:0.029]; disease duration was not different. CVID pts with AID had a higher familiarity for AID (23.9% vs 5.4%; p:0.032). CVID pts with AID had less gastro-enteric infections (21.7% vs 45.9%; p:0.033) and chronic sinusitis (28.3% vs 54.1%; p:0.024), whereas lung damage as bronchiectasis was not different among the two groups. Inflammatory bowel disease like diagnosis was more frequent in pts with AID (15.2% vs 0%; p: 0.015). No differences among granuloma, hepato-spleen axis abnormalities, neoplasia were found. CVID patients with AID received more frequently IS treatment for autoimmunity, enteropathy or granuloma (65.2% vs 5.4%; p:<0.001). Autoimmune cytopenia was the most common autoimmune manifestation occurring in 19 (22.9%) of the pts, with immune thrombocytopenia being the most prevalent. IS treatment was necessary in 17/19 (89.5%) of the pts affected by autoimmune cytopenia; in all 17 pts high-dosage steroid therapy was employed, while in 5 cases, due to refractory cytopenia, rituximab was used achieving persistent remission in 80.0% of the pts. A diagnosis of IRD was made in 12/83 (13.3%) pts (table 1). Among the 12 pts affected by IRD, 9/12 (75.0%) suffered from arthritis, being in two cases related to a diagnosis of Behçet’s Disease, in 4 cases to a diagnosis of Psoriatic Arthritis, in one case Undifferentiated Spondyloarthritis and in one case Enteropathic Arthritis. Genetic analysis searching for mutations potentially leading to CVID clinical picture had been performed in 31/83 (37.3%) pts; analysis resulted positive in 41.9% of the tested patients. Genetic variants potentially linked to inborn errors of immunity (IEI) were found in more than half of CVID pts with AID, much more frequently than in other CVID patients [13/23 (56.5%) vs 0/8 (0.0%); p:0.010]. In one pt with a clinical diagnosis of Behçet’s Disease, NGS analysis identified two variants, one pathogenic and the other likely pathogenic, on the alleles of the gene Adenosine Deaminase 2, aiding in suspending the on-course inefficient IS treatment and substituting it with an anti-TNF alfa therapy (Table 1), leading to remission of the IRD. The identified genetic variants are described in Table 2.
Conclusion: CVID can be complicated by a wide spectrum of clinical pictures beside recurrent infections. IRD may contribute to complicate this condition, in our cohort being in most of the cases inflammatory arthritis, which could be categorized as a form of Spondyloarthritis in about two third of the cases, requiring IS treatment for the rheumatic condition itself or due to other associated granulomatous or AID. Notably, CVID pts affected by AID and IRD showed a higher frequency of genetic variants potentially leading to IEI; in one case genetic testing aided in orienting IS treatment, leading patient to remission of the IRD. Nowadays genetic analysis and the identification of monogenic variants has still limited implications in influencing possible treatments in CVID, but in the future it might help in targeting precise mechanisms to treat patients with AID and IRD.
REFERENCES: [1] Thalhammer J et al, J Allergy Clin Immunol 2021
[2] Maffucci P et al, Front Immunol 2016
[3] Seidel MG et al, J Allergy Clin Immunol 2019
Acknowledgements: NIL.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (