fetching data ... 
Background: Adult-onset Still’s disease (AOSD) is a chronic and systemic autoinflammatory disorder, with the possibility to result in life-threatening complications, including macrophage activation syndrome (MAS). The metabolic-immune interplay underlying the immunopathology of AOSD/MAS remains largely unexplored.
Objectives: To explore monocyte/macrophage-derived immunomodulatory metabolite itaconate in hepatic immunopathology via CXCL10-CD8 T-cell axis in AOSD.
Methods: Various methods including metabolomics, ELISA, western blotting, qRT-PCR, flow cytometry, and immunohistochemistry were employed to analyze samples from patients, animals and cell cultures. A CpG ODN 1826-induced MAS mouse model was used for in vivo disease manifestation. The Acod1 -deficient mice was employed to address the Acod1-itaconate axis.
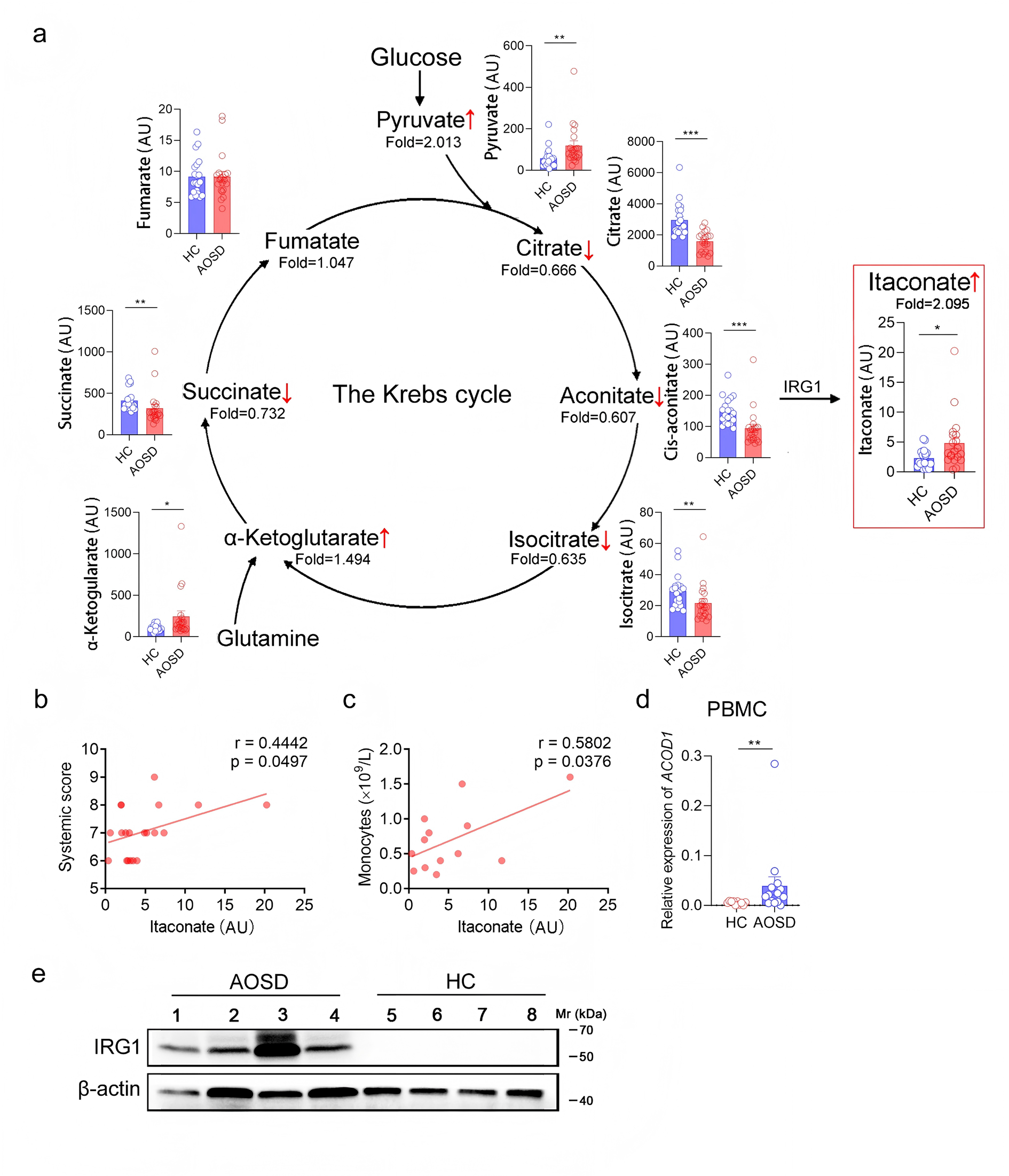
Results: Clinical metabolomics revealed itaconate— a myeloid cell-specific metabolite derived from tricarboxylic acid (TCA) cycle via the enzyme aconitate decarboxylase 1 (ACOD1)— was elevated in sera of AOSD patients, attributable to peripheral blood monocytes and correlated with disease severity. This was consolidated by the identification of Acod1 -itaconate axis in monocytes and macrophages in both mouse model of MAS and in vitro cell cultures. While itaconate suppressed IL-1β, IL-6, CXCL1 and CCL2, it paradoxically amplified CXCL10 secretion in macrophages. This was in line with the observation of elevated plasma CXCL10 level in MAS patients. In the MAS mouse model, ablation of Acod1 ameliorated disease manifestations and hepatic inflammation, accompanied by a reduced CXCL10 level and attenuated hepatic infiltration of CD8+ T cells.
Conclusions: Our study reveals a previously unrecognized metabolic-immune crosstalk in AOSD/MAS, positioning monocyte/macrophage-derived itaconate as a dual regulator that suppresses canonical proinflammatory cytokines whereas licensing CXCL10-mediated CD8+ T cell-driven tissue injury.Therefore, discovery from this study calls for scrutiny of itaconate-based anti-inflammatory strategies in chronic inflammatory diseases.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.