fetching data ... 
Background: Mixed connective tissue disease (MCTD) is a rare autoimmune condition, defined by U1RNP-antibodies and overlapping clinical features of multiple connective tissue diseases. Although myositis is a recognized manifestation of MCTD, the morphological characteristics of U1RNP-positive myositis, as well as the underlying pathomechanisms driving disease development, remain poorly understood [1]. On the other hand, subclassification of myositis patients can offer new pathways for better diagnosis and patient management, making the morphological analysis as well as distinct pathophysiological aspects crucial for further potential classifications.
Objectives: The aim of this study was to perform an in depth histopathological and transcriptional analysis of U1RNP-positive myositis to uncover possible pathomechanisms and/or distinct phenotypes.
Methods: 20 muscle biopsies from anti-U1RNP-positive patients presenting with clinical and morphological features of myositis were analyzed by immunohistochemistry and transcriptomic studies. Clinical data were analyzed retrospectively. Findings were put in context with biopsies of other myositis entities and non-disease controls.
Results: The majority of patients were female (75%, 15/20), with a mean age of 49 years (range 24–72). Most patients (94%, 16/17) presented with myalgia and proximal muscle weakness. Arthralgia was observed in 73% (11/15), Raynaud’s phenomenon in 75% (6/8), interstitial lung disease in 30% (3/10), and cardiac involvement in 30% (3/10). Antinuclear antibodies were positive in all patients. Concomitant autoantibodies were detected in 94% (16/17) of cases, most commonly anti-SSA, anti-CCP antibodies, or rheumatoid factor.
Histopathologic analysis indicated three distinct subtypes of U1RNP-positive myositis: 30% (6/20) of the biopsies showed features resembling anti-synthetase syndrome (ASyS) with perifascicular MHC-I and II positivity on myofibers, infiltrates and necrosis. Additionally, these muscle specimens demonstrated signs of perivasculitis. 45% (9/20) of the examined biopsies displayed features consistent with immune-mediated necrotizing myopathy (IMNM), including necrosis and myophagocytosis and additionally exhibited MHC-II positivity in a diffuse, focally enhanced pattern. But these biopsies lacked the typical IMNM-like p62 pattern. Moreover, 25% (5/20) of the biopsies demonstrated minimal myositis with capillary pathology-like pattern (a common feature of scleromyositis (SM)). This entity also exhibited a mild myofiber necrosis in 80% and a perifascicular pattern in 40% of the cases.
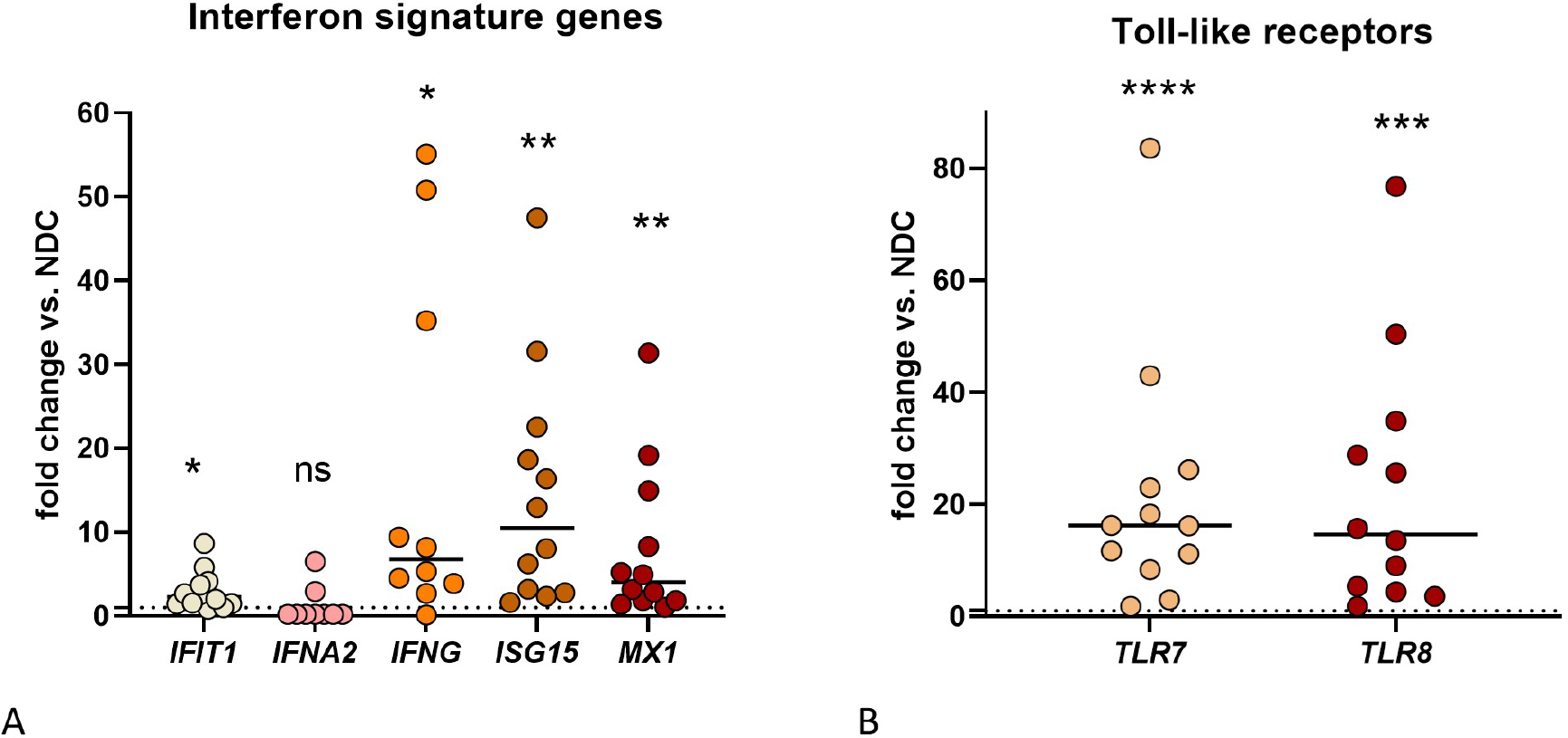
Transcriptional results using quantitative polymerase chain reaction from 13 U1RNP-positive biopsies showed a significantly increased expression of interferon-associated genes (see Figure 1A), with the most prominent type 1 and type 2 interferon responses in IMNM-like biopsies (e.g. IFNG, ISG15, MX1 ). Furthermore, we detected a strongly increased gene expression of toll-like receptors TLR7 and TLR8 compared to non-disease controls (see Figure 1B).
Conclusions: This study revealed three subtypes of U1RNP-positive myositis, resembling, yet substantially differing from other myositis entities like ASyS, IMNM and SM. Transcriptomic results showed significantly increased toll-like receptors and interferon genes, the latter with a prominent expression especially in the IMNM-like subgroup.
These findings raise the question whether U1RNP-positive myositis is one disease entity in the context of MCTD or rather reflects a complex overlap of various pathophysiological mechanisms. Further research is planned to unravel these differences.
REFERENCES: [1] Muscular and extramuscular features of myositis patients with anti-U1-RNP autoantibodies. M. Casal-Dominguez et al. Neurology. 2019. DOI:10.1212/WNL.0000000000007188
Acknowledgments: NIL.
Disclosure of Interests: None declared.