fetching data ... 
Background: The immunobiology of flare versus maintenance of remission in rheumatoid arthritis (RA) is poorly understood, including any role of monocytes. BIO-FLARE was a prospective multicentre experimental medicine study in which patients with RA in remission (DAS28-CRP <2.4) on csDMARDs stopped all RA treatment and were followed up for 24 weeks, or until flare [1]. Here we report the results of comprehensive transcriptomic and epigenomic (chromatin accessibility) profiling of peripheral blood monocytes from the BIO-FLARE study.
Objectives: To explore the mechanistic role of monocytes in predisposing to or driving flare by assessing differences in gene expression and chromatin accessibility at baseline and over time among BIO-FLARE participants who flared or remained in remission.
Methods: Flare was defined as DAS28-CRP ≥3.2 or ≥2.4 twice within two weeks. As per the previously published clinical data, approximately 50% of 121 BIO-FLARE participants experienced flare, while 50% remained in remission [2]. In these analyses, only participants with paired baseline and follow-up data were included. Baseline visits were the date of csDMARD cessation. Follow-up visits were the flare visit for those who flared, or the final (week 24) visit for those who remained in remission. Pre-flare visits (the most recent study visit prior to flare) were also included. Bulk RNA-seq and ATAC-seq were performed on cryopreserved isolated peripheral blood CD14+ cells, with 150 base-pair paired-end sequencing at target depths of 30 and 50 million reads, respectively. Pre-processing, alignment, consensus peak calling (for ATAC-seq), and QC were performed according to established pipelines. Differential analyses were conducted using DESeq2, with significance thresholds of absolute log2FC >0.5 and FDR adjusted p-value <0.05.
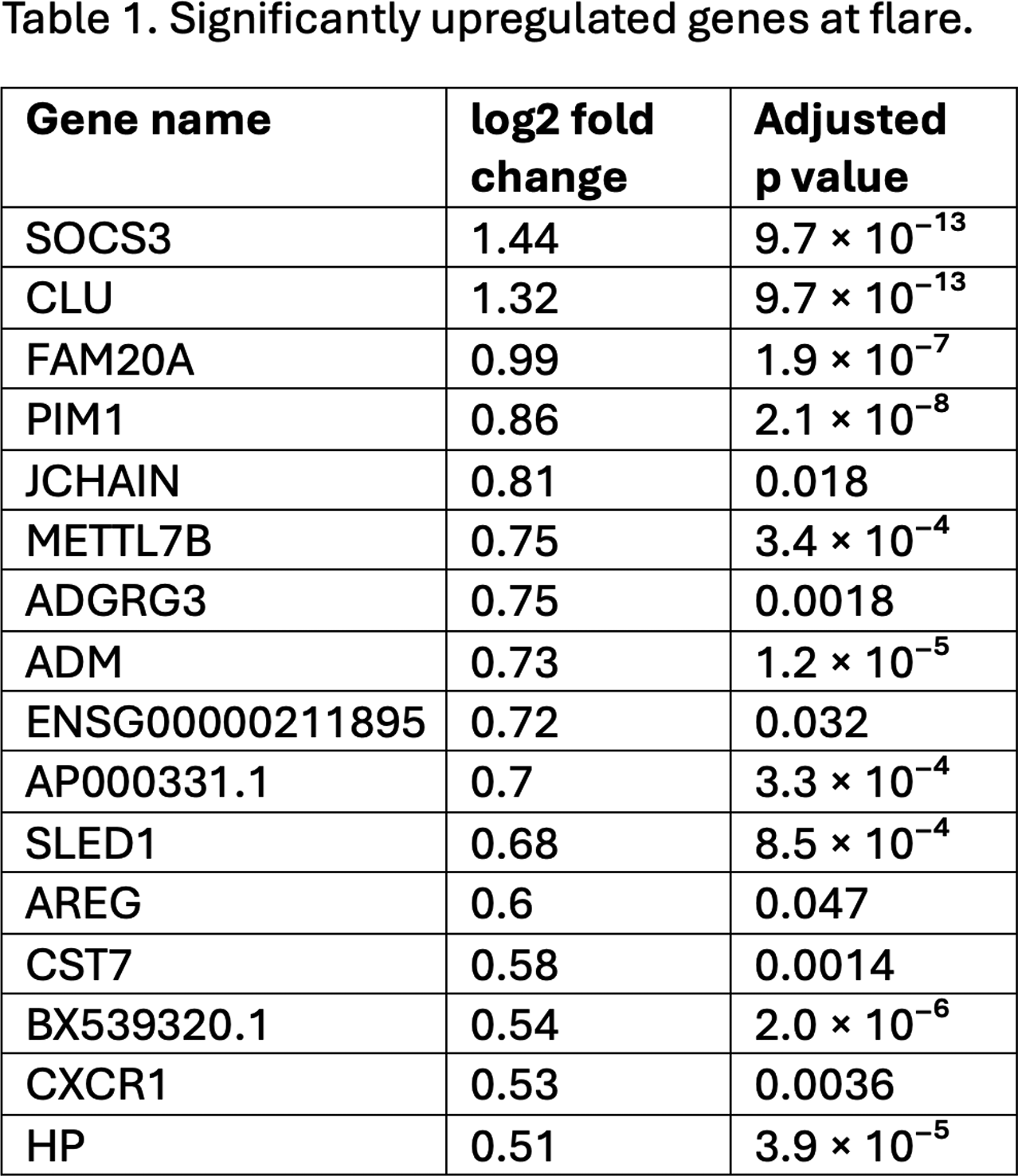
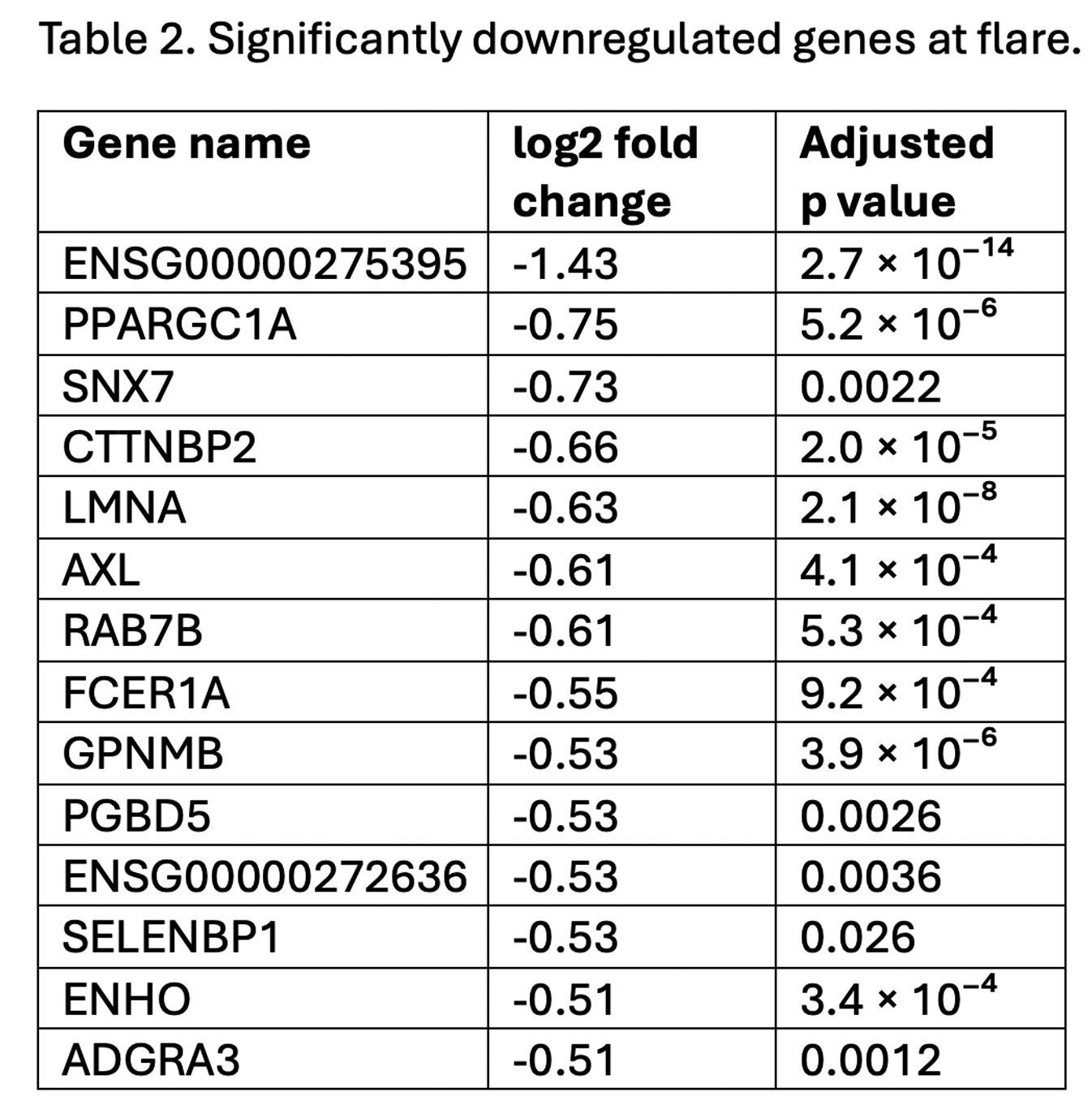
Results: A total of 222 samples from 90 BIO-FLARE participants (of whom 47 flared and 43 remained in remission) were included. Among participants who flared, RNA-seq analysis revealed 30 differentially expressed genes (DEGs) at flare versus baseline, consisting of 16 upregulated and 14 downregulated genes (tables 1 and 2). The DEG profile at flare included genes implicated in IL6-pSTAT3 signalling ( SOCS3 , PIM1 ), pro-inflammatory polarisation ( AXL , RAB7B , FCER1A , GPNMB ), suppression of apoptosis ( PIM1 , CLU , AREG ), enhanced migratory capacity ( CXCR1 , LMNA ), and associated metabolic reprogramming favouring glycolysis ( PPARGC1A ). When an alternative flare definition requiring objective signs of inflammation was applied (increase in both swollen joint count ≥2 and CRP ≥4mg/L), 101 DEGs were identified, including many with similar putative functions. The DEG profile seen at flare was not seen in advance of flare when comparing pre-flare visits [median (IQR) 16 (11-25) days before flare] to baseline. Minimal DEGs were identified at baseline between participants who subsequently flared versus those who remained in remission, or among remission participants between week 24 versus baseline. ATAC-seq analysis identified no significant differences in chromatin accessibility across any comparison.
Conclusions: Based on this multi-omic analysis, circulating monocytes do not appear primed at the transcriptomic or chromatin accessibility level towards flare following csDMARD cessation. A distinct pro-inflammatory DEG profile was seen at flare, but not in advance of flare, suggesting monocytes may be responders rather than key early initiators. Dynamic changes in chromatin accessibility do not seem to play a role in orchestrating these changes in gene expression.
REFERENCES: [1] Rayner et al, BMC Rheumatol, 2021.
[2] Rayner et al, BMJ Open, 2025.
Acknowledgments: NIL.
Disclosure of Interests: Andrew Melville MRC GSK EMINENT PhD fellowship (2020-2024), Kieran Woolcock: None declared, Fiona Rayner: None declared, Andrew McGucken: None declared, Amy E Anderson: None declared, Andrew Filer Speaker/consulting fees from Roche, Janssen and Sonoma, Institutional research grants from BMS, Roche, UCB, Nascient, Mestag, GSK, Janssen, Iain B. McInnes Honoraria or research support from Abbvie, Janssen, Novartis, Eli Lilly, Astra Zeneca, GSK, BMS, Moonlake, Evelo, Causeway THerapeutics, Cabaletta, Roche, Pfizer and Compugen, Karim Raza Personal fees for lectures/consultancy from Abbvie and Sanofi, Research grant support from Bristol Myers Squibb, Arthur Pratt: None declared, Kenneth F Baker Consulting fees from Modern Biosciences, Research support from Genentech, clinical improvement funding from Pfizer, John Isaacs Consulted for Abbvie, Anaptys Bio, AstraZeneca, BMS, Eli Lilly, Galapagos NV, Gilead Sciences Ltd, GSK, Istesso Ltd, Kira Biotech, Ono Pharma, Pfizer, Revelo Biotherapeutics, Roche and Sanofi, Research grants from Pfizer, Janssen and GSK, Stefan Siebert Amgen, Johnson & Johnson, Novartis, Pfizer and UCB, AbbVie, AstraZeneca, Johnson & Johnson, Syncona, Shattuck Labs, Teijin Pharma, and UCB, Eli Lilly, GlaxoSmithKline, Johnson & Johnson, Pfizer and UCB, Carl S Goodyear: None declared.