fetching data ... 
Background: Rheumatoid arthritis (RA) is characterised by persistent synovial inflammation and progressive bone erosion driven by excessive osteoclast activity. Although contemporary disease-modifying antirheumatic drugs effectively suppress immune-mediated inflammation, structural joint damage frequently continues, indicating that osteoclast-intrinsic mechanisms remain insufficiently targeted. Thromboxane A2 (TXA2), a bioactive lipid derived from arachidonic acid metabolism, has been implicated in pathological bone resorption. However, its contribution to osteoclast metabolic programming and inflammatory bone destruction in RA remains poorly defined.
Objectives: To investigate the role of thromboxane A2 receptor (TBXA2R) signalling in osteoclast differentiation and inflammatory bone erosion in RA, and to elucidate the metabolic mechanisms underlying this process.
Methods: Systemic and local TXA2 biosynthesis was assessed in patients with RA, osteoarthritis, and healthy controls by quantifying urinary 11-dehydro-TXB2 and synovial TXB2. Osteoclast differentiation from murine bone marrow–derived macrophages was examined using TXA2 mimetics, including carbocyclic thromboxane A2 (CTA2) and U46619, and pharmacological TBXA2R antagonists, including seratrodast (STD) and SQ29548 (SQ). Transcriptional and metabolic reprogramming was characterised using single-cell RNA sequencing, bulk transcriptomics, targeted metabolomics, and Seahorse-based mitochondrial respiration assays. In vivo relevance was evaluated using collagen-induced arthritis (CIA) models following pharmacological TBXA2R inhibition with STD or SQ, or genetic deletion of TBXA2R, combined with histology and micro–computed tomography analyses.
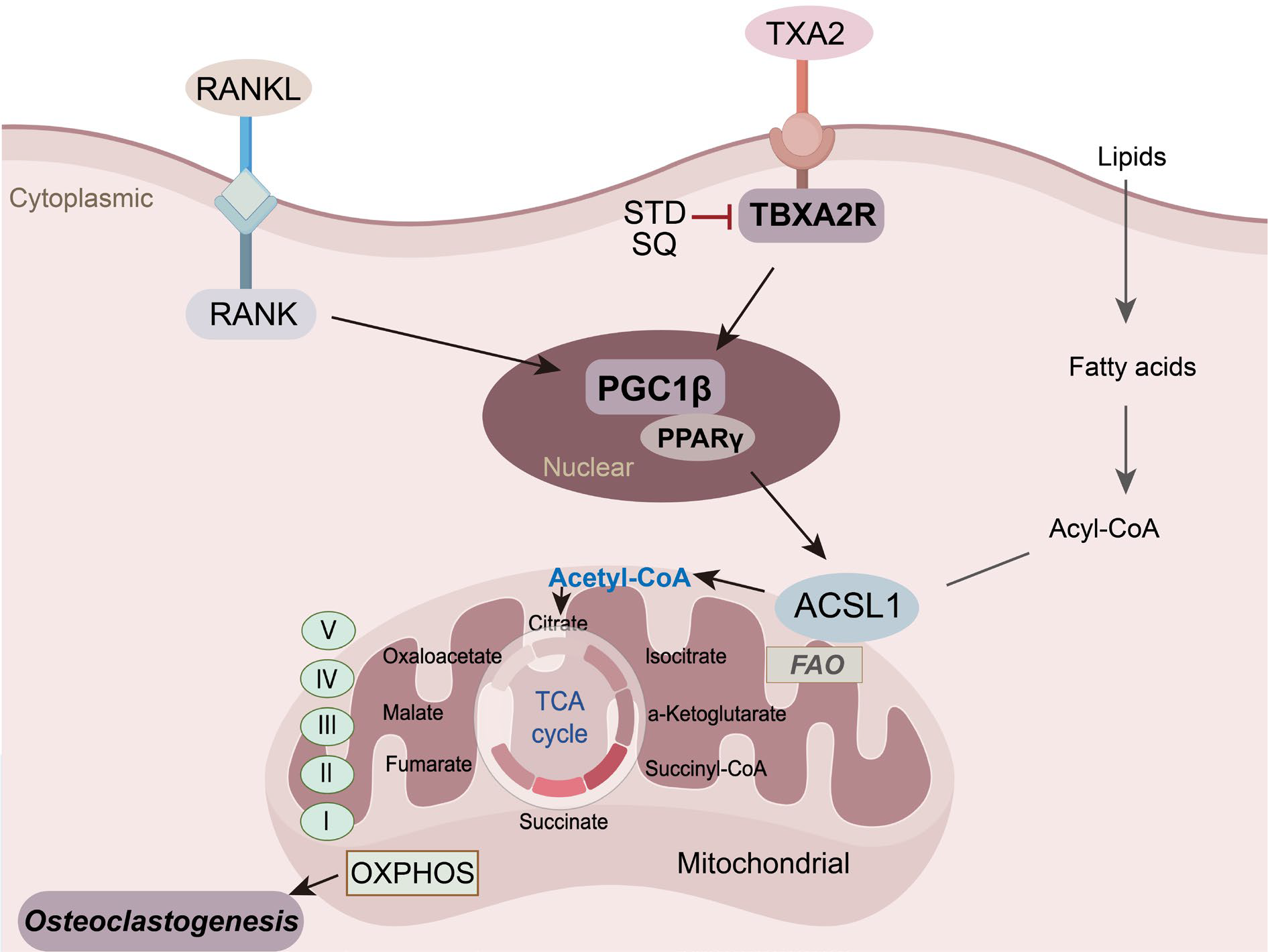
Results: TXA2 biosynthesis was significantly elevated in RA patients both systemically and within inflamed joints. TXA2 signalling synergised with RANKL to promote osteoclast differentiation, whereas pharmacological blockade of TBXA2R markedly suppressed osteoclast formation without affecting cell viability. Single-cell transcriptomic analysis revealed that TBXA2R inhibition redirected osteoclast precursors away from a pro-osteoclastogenic trajectory and was associated with suppression of oxidative phosphorylation and tricarboxylic acid cycle pathways. Integrated metabolomic profiling identified fatty acid oxidation as the dominant source of acetyl-CoA fuelling mitochondrial respiration during osteoclastogenesis, mediated by acyl-CoA synthetase long-chain family member 1 (ACSL1). Mechanistically, TBXA2R signalling maintained ACSL1 expression through activation of the PGC-1β/PPARγ transcriptional axis, thereby sustaining mitochondrial respiration. In CIA mice, pharmacological or genetic inhibition of TBXA2R reduced osteoclast accumulation, preserved trabecular microarchitecture, and maintained bone mineral density.
Conclusions: TBXA2R signalling promotes inflammatory bone erosion in RA by sustaining ACSL1-dependent fatty acid oxidation and mitochondrial respiration in osteoclasts. Disruption of this metabolic programme through TBXA2R inhibition protects against arthritis-associated bone loss, identifying TBXA2R as a metabolically defined therapeutic target for preventing structural joint damage in RA.
Mechanistic model illustrating how TBXA2R signalling amplifies RANKL-driven osteoclastogenesis by sustaining ACSL1-mediated fatty acid oxidation and oxidative phosphorylation.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.