fetching data ... 
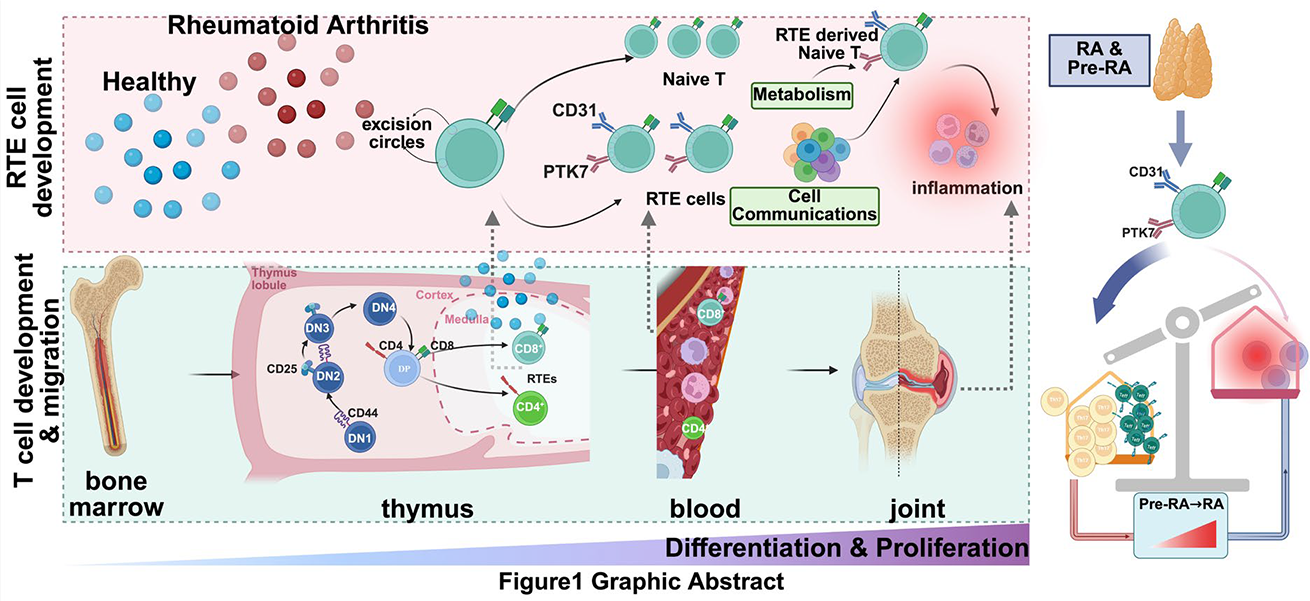
Background: Recent thymic emigrants (RTEs) constitute the earliest naïve T-cell compartment exported from the thymus, yet their role in rheumatoid arthritis (RA) remains elusive because inflammatory milieus can distort commonly used phenotypic definitions [1-3]. Whether bona fide RTEs undergo an early, disease-relevant remodeling during the transition from pre-clinical autoimmunity to established RA, and whether such remodeling mechanistically links systemic immunity to synovial inflammation, is unknown.
Objectives: We aimed to establish an inflammation-robust definition of bona fide RTEs, map their state transitions across the pre-clinical to active RA continuum, determine whether a conserved immunometabolic program defines RTE deviation in RA, and dissect its tissue origin (peripheral conditioning versus altered thymic output) and cross-tissue coupling with synovial inflammatory circuits. We further used PD-1 blockade–associated inflammatory arthritis and chronic HBV as outcome-divergent comparators to distinguish acute checkpoint-driven remodeling and tolerance-biased states from RA-specific chronic locking.
Methods: We developed a composite RTE definition[1,2] anchored to thymic recency using orthogonal validation and applied it to large-scale immune profiling spanning pre-RA and RA, complemented by synovial datasets and targeted experimental validation. Circulating inflammatory context, cross-cell interaction programs, functional metabolic phenotyping, controlled tissue-mimicking co-cultures, and time-resolved arthritis modeling were integrated to establish mechanistic directionality and disease-stage dependencies.
Results: We defined a transferable bona fide RTE framework [4] that remained stable under inflammatory conditions and showed that conventional single-marker strategies can misclassify inflammation-conditioned naïve T cells as RTEs. Across the pre-RA to RA continuum, bona fide RTEs displayed an early and persistent immunometabolic deviation dominated by glycolysis, accompanied by reduced identity stability and biased fate trajectories, marking a discrete metabolic activation threshold that we term a glycolytic gate. This glycolytic RTE state emerged before overt disease and became reinforced in established RA, consistent with progressive immunological locking. Cross-disease analyses separated shared inflammatory components from disease-specific programs: PD-1 blockade–associated arthritis exhibited an acute checkpoint-perturbed RTE remodeling, whereas chronic HBV preferentially aligned with tolerance-associated features rather than the RA-like glycolytic gate and fate drift. Importantly, the glycolytic RTE state was coupled to synovial inflammatory and stromal circuits, supporting a cross-tissue feed-forward model. Functional assays confirmed that the transcript-inferred glycolytic shift reflected bona fide metabolic rewiring, and controlled perturbations attenuated the glycolytic gate and associated fate deviation. Time-resolved modeling further indicated stage-dependent contributions of peripheral inflammatory conditioning and altered thymic output, providing a mechanistic explanation for early vulnerability and later persistence.
Conclusions: Our study establishes a robust definition of bona fide RTEs under inflammation and identifies an RA-associated glycolytic gate that emerges in pre-clinical disease, couples blood RTE states to synovial inflammatory circuits, and becomes chronically reinforced during progression. By leveraging PD-1 blockade and chronic HBV as human benchmarks for checkpoint perturbation and tolerance bias, we disentangle acute inflammatory remodeling from RA-specific chronic locking, positioning RTE immunometabolic programming as an actionable axis for early stratification and mechanism-guided intervention[5].
REFERENCES: [1] Haines CJ, et al. J Exp Med 2009.
[2] Bains I, et al. PLoS One 2013.
[3] Bohacova P, et al. Immunity 2024.
[4] Kraus FV, et al. Ann Rheum Dis 2022 Suppl.
[5] Krienke C, et al. Science 2021.
Acknowledgments: NIL.
Disclosure of Interests: None declared.