fetching data ... 
Background: Ankylosing spondylitis (AS) is a chronic inflammatory autoimmune disorder characterized by progressive spinal stiffness, pain, and functional impairment, leading to substantial deterioration in patients’ quality of life. Accumulating epidemiological evidence in recent years suggests that metabolic factors may be involved in the pathophysiology of AS. Specifically, observational studies have shown that individuals with AS frequently exhibit multiple abnormal metabolic traits (MTs)[1-3]. However, these observed associations are inherently constrained by residual confounding, lifestyle-related influences, medication use, and the possibility of reverse causation, thereby precluding definitive conclusions regarding whether metabolic abnormalities causally contribute to AS or instead represent downstream consequences of the disease.
Objectives: Therefore, this study aimed to investigate the causal relationships between MTs and AS using Mendelian randomization analysis.
Methods: Using publicly available large-scale genome-wide association study (GWAS) summary statistics, we systematically investigated the potential causal relationships between ankylosing spondylitis and a comprehensive panel of metabolic phenotypes. This panel comprised 22 metabolic traits, including anthropometric measures, lipid-related traits, glycaemic-related traits, blood pressure traits, liver and kidney function markers, and inflammatory or metabolic biomarkers (e.g., C-reactive protein [CRP] and serum uric acid [SUA]). In addition, six metabolic diseases were examined: coronary artery disease (CAD), heart failure (HF), type 2 diabetes (T2D), hypertension, non-alcoholic fatty liver disease (NAFLD), and chronic kidney disease (CKD). MR was adopted as the primary analytical framework, leveraging genetic variants as instrumental variables to strengthen causal inference by reducing confounding and reverse causation. To address additional biases inherent to summary-level MR analyses—particularly those related to weak instruments, winner’s curse, and sample overlap between exposure and outcome GWAS—we implemented MRlap as the main MR approach [4]. MRlap refines conventional inverse-variance weighted (IVW) estimates by explicitly modeling the genetic architecture of the traits and incorporating parameters such as exposure heritability, polygenicity, and cross-trait linkage disequilibrium score intercepts. In the absence of a statistically significant difference between the IVW and MRlap-corrected estimates ( P > 0.05), the IVW result was considered robust and retained. Conversely, when a significant discrepancy was observed ( P < 0.05), the MRlap-corrected estimate—less susceptible to bias from sample overlap—was prioritized as the primary causal inference. To control for multiple testing, statistical significance was determined using a Bonferroni-corrected threshold of P < 8.93×10 -4 (0.05 divided by 28 metabolic phenotypes and two MR directions).
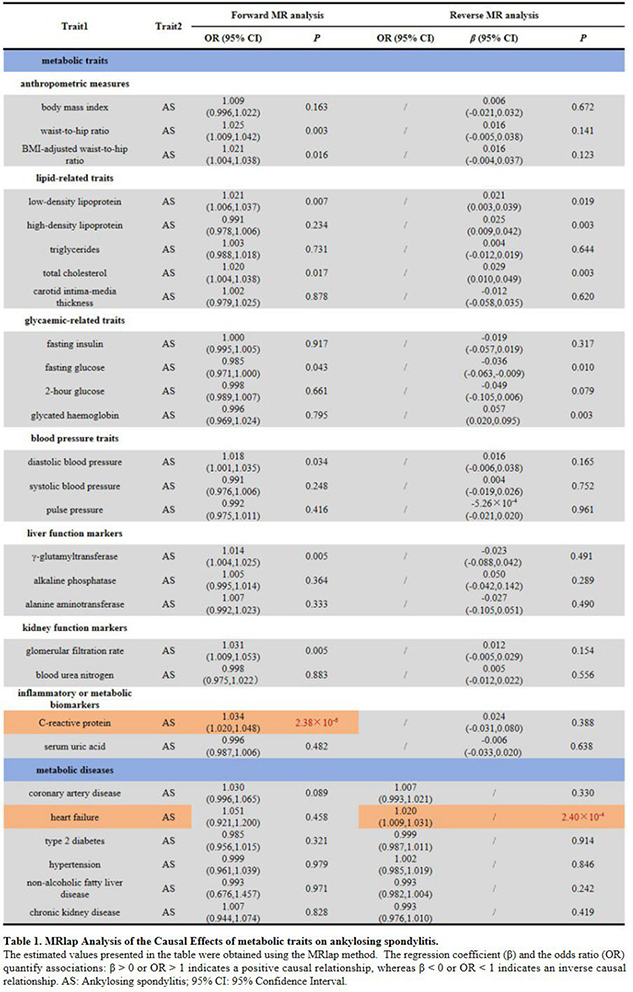
Results: In forward MR analyses, CRP was the only metabolic phenotype showing a significant positive causal association with AS (OR: 1.034, 95% CI: 1.020–1.048; P = 2.38×10 -6 ), with each one–standard-deviation increase in genetically proxied CRP corresponding to an approximately 3.4% higher odds of AS. This finding supports a contributory role of genetically determined systemic inflammatory liability in AS susceptibility, rather than CRP merely reflecting downstream disease activity. In contrast, reverse MR analyses identified a significant positive causal association only between genetic liability to AS and HF (OR: 1.020, 95% CI: 1.009–1.031; P = 2.40×10 -4 ), indicating that AS may causally increase the risk of HF.
Conclusions: Collectively, these results support an asymmetric causal relationship in which systemic inflammatory liability contributes to AS susceptibility, whereas AS may confer increased downstream cardiovascular risk.
REFERENCES: [1] Kang, K. Y., Hong, Y. S., Park, S. H. & Ju, J. H. Low levels of serum uric Acid increase the risk of low bone mineral density in young male patients with ankylosing spondylitis. J Rheumatol 42 , 968-974, doi:10.3899/jrheum.140850 (2015).
[2] Robinson, A. C., Teeling, M. & Casey, E. B. Hepatic function in ankylosing spondylitis. Ann Rheum Dis 42 , 550-552, doi:10.1136/ard.42.5.550 (1983).
[3] Liao, K. F., Kuo, Y. H. & Lai, S. W. Diabetes mellitus in ankylosing spondylitis. Ann Rheum Dis 80 , e134, doi:10.1136/annrheumdis-2019-216221 (2021).
[4] Mounier, N. & Kutalik, Z. Bias correction for inverse variance weighting Mendelian randomization. Genet Epidemiol 47 , 314-331, doi:10.1002/gepi.22522 (2023).
Acknowledgments: NIL.
Disclosure of Interests: None declared.