fetching data ... 
Background: Mesenchymal stromal cells (MSCs) are key players in regenerative medicine, supporting hematopoiesis, tissue repair, and immune modulation. Their ability to home to inflamed sites makes them attractive candidates for treating inflammatory diseases and tissue injury. In systemic lupus erythematosus (SLE), MSCs improve disease outcomes in animal models and patients, enhancing apoptotic cell clearance and dampening T-cell responses with few side effects. However, clinical trials have produced inconsistent results. Variability in donor characteristics (age, sex, genetics, health), cell source (autologous vs. allogeneic), isolation and culture conditions, cryopreservation/thawing protocols, and delivery route can alter MSC function. Moreover, hostile inflammatory microenvironments and concomitant drugs may blunt their therapeutic efficacy, especially in autologous settings where an autoimmune milieu could further impair MSC activity.
Objectives: These variables underscore the need to understand how chronic inflammatory autoimmune microenvironments modulate MSC biology and function to improve therapeutic consistency.
Methods: To reveal how chronic autoimmune inflammation reshapes MSC biology and function, we compared bone-marrow-derived MSCs (BM-MSCs) from lupus-prone mice harvested during the pre-clinical (healthy) versus the established disease phase. Cells were characterized by flow cytometry for canonical MSC- and senescence-associated markers. Bulk RNA-Seq was performed to delineate transcriptomic signatures and functional pathways. Secreted cytokines and chemokines were quantified with multiplex bead-based arrays. In vitro differentiation assays were employed to assess multipotency, while co-culture with immune cells was used to evaluate immunomodulatory capacity.
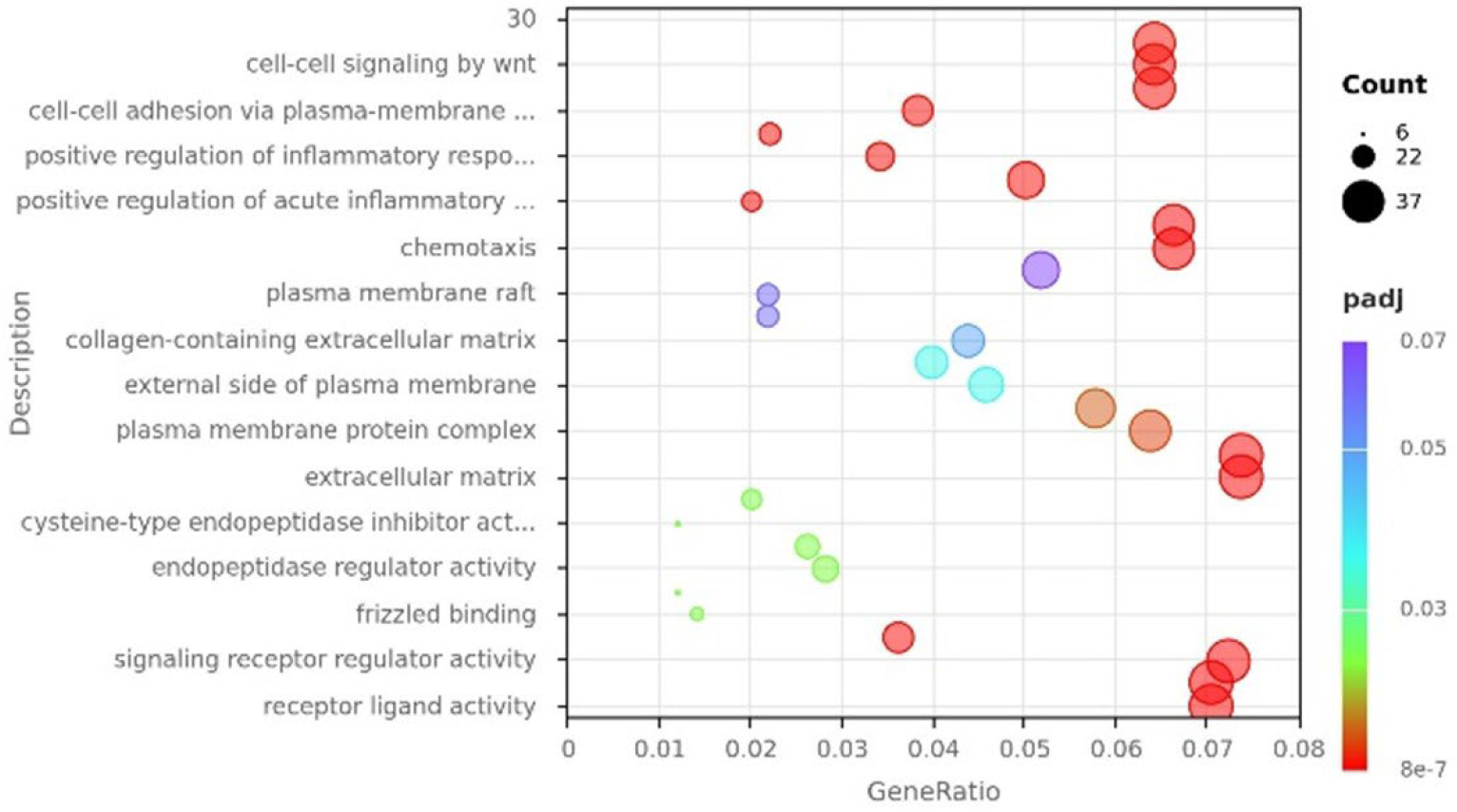
Results: Our preliminary data showed no differences in the expression of canonical MSC markers, which overall displayed heterogeneous expression patterns consistent with the presence of subpopulations. However, transcriptomic analysis by RNA sequencing identified a total of 401 differentially expressed genes (DEGs) in MSCs derived from sick NZB/W F1 mice compared with MSCs from healthy NZB/W F1 controls (log2FC ≥ 1, p-value ≤ 0.05). Gene ontology analysis (GO) of upregulated genes showed enrichment of genes associated with chemotaxis, cell adhesion, and positive regulation of inflammatory response, among others. To assess the biological relevance of transcriptional changes, gene set enrichment analysis (GSEA) was performed. Gene sets related to stem cell pluripotency, signaling, and metabolic pathways, including mTOR, PPAR, WNT, and OXPHOS, as well as pathways involved in cellular stress responses, were differentially enriched. These findings suggest a potential functional impairment of MSCs exposed to an inflammatory microenvironment. Despite these transcriptomic alterations, staining for canonical senescence-associated markers (γH2AX, β-galactosidase) and the proliferation marker Ki67 revealed no significant differences between MSCs from sick and healthy NZB/W F1 mice. Nonetheless, a modest reduction in colony-forming unit (CFU) capacity, osteogenic potential and increased production of certain inflammatory cytokines were observed, but only in late-passage cells. Importantly, MSCs from both healthy and sick NZB/W F1 mice retained their immunomodulatory capacity, effectively modulating macrophage polarization and T cell proliferation.
Conclusions: Chronic autoimmune inflammation induces subtle molecular and phenotypic changes in MSCs; however, their core functional properties, particularly immunomodulatory capacity, are largely preserved.
Transcriptional reprogramming of bone marrow-derived MSCs in late-stage lupus. Gene-ontology analysis of genes up-regulated in MSCs isolated from lupus-prone, nephritic mice relative to pre-disease controls.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.