fetching data ... 
Background: Monogenic immune dysregulation, classified under inborn errors of immunity, frequently manifests in childhood as severe, multisystem autoimmunity and/or autoinflammation, often mimicking refractory rheumatic disease. While genomic diagnostics increasingly enable precise identification of the underlying pathway defects, translation of these findings into effective, pathway-aligned targeted therapy remains a major challenge in resource-limited settings due to delayed diagnosis, limited availability, and financial barriers.
Objectives: To assess outcomes of genotype-guided, pathway-aligned therapy in pediatric monogenic immune dysregulation and to examine the effect of diagnostic delay and access barriers on response.
Methods: This is single-centre retrospective non-intervention observational study based on anonymized clinical and genetic data included children with disease onset <18 years presenting with severe autoimmune or autoinflammatory phenotypes. All patients were evaluated and followed at this centre and underwent clinical or whole exome sequencing. Variants were considered actionable if classified as pathogenic/likely pathogenic, or if a phenotype-concordant VUS fulfilled a structured 9-point AI-assisted genotype–phenotype concordance framework incorporating gene–disease association, functional domain involvement, evolutionary conservation, predicted amino acid impact, population allele frequency, in silico pathogenicity prediction, clinical phenotype match, segregation data and available functional evidence. Based on the inferred dysregulated immune pathway, pathway-aligned targeted therapy was selected. Outcomes were classified as complete response (inactive disease ≥1 month off systemic glucocorticoids), partial response (improvement with steroid taper) or persistent refractory/relapse. Secondary analyses assessed the impact of diagnostic delay and access limitations on response.
Results: The cohort comprised 25 children (male:female 18:7). Median age at disease onset was 1.5 years (IQR 0.5–5 ) and median age at genetic diagnosis 6.0 years (IQR 3–10 ), with a mean diagnostic delay of 4.8±3.2 years . Predominant phenotypes were autoimmune in 17/25 (68% ), autoinflammatory in 3/25 (12% ), mixed autoimmune–autoinflammatory in 3/25 (12% ) and hyperinflammation in 2/25 (8% ). Major manifestations included arthritis 10/25 (40% ), enteropathy/colitis 8/25 (32% ), SLE/lupus-like disease 5/25 (20% ), autoimmune cytopenias 5/25 (20% ), and interstitial lung disease, necrotising/granulomatous inflammation, CNS inflammation and systemic vasculitis each in 3/25 (12% ); endocrinopathy (thyroiditis/T1DM) occurred in 2/25 (8% ). Recurrent/severe infections were present in 11/25 (44% ), refractory allergy in 4/25 (16% ) and lymphoproliferation in 1/25 (4% ).
Immunological evaluation showed abnormal immunoglobulin profiles in 16/23 (69.6% ) tested (elevated in 47.8%, low in 21.7%), deranged lymphocyte subsets in 5/10 (50% ) tested, and positive autoantibodies in 11/23 (47.8% ).
Genetic analysis revealed marked heterogeneity with 23 disease-associated genes across 25 children (MVK and NOD2 each 8%, and single cases of ADAM17, BTK, C1Q, C1QB, COPA, CTLA4, CYBB, DOCK8, FCGR2B, IKBKB, LRBA, PLCG1, PSMB8, RAG1, RELA, RNF31, RUNX1, SOCS1, STXBP2, TCF3 and WAS). Primary variants were pathogenic in 9/25 (36% ), likely pathogenic in 7/25 (28% ) and VUS in 9/25 (36% ), with stepwise separation of 9-point genetic scores (VUS median 7.0, IQR 6.5–7.5, range 5.0–8.5 ; likely pathogenic median 7.5, IQR 7.0–7.5 ; pathogenic median 8.0, IQR 8.0–8.0 ).
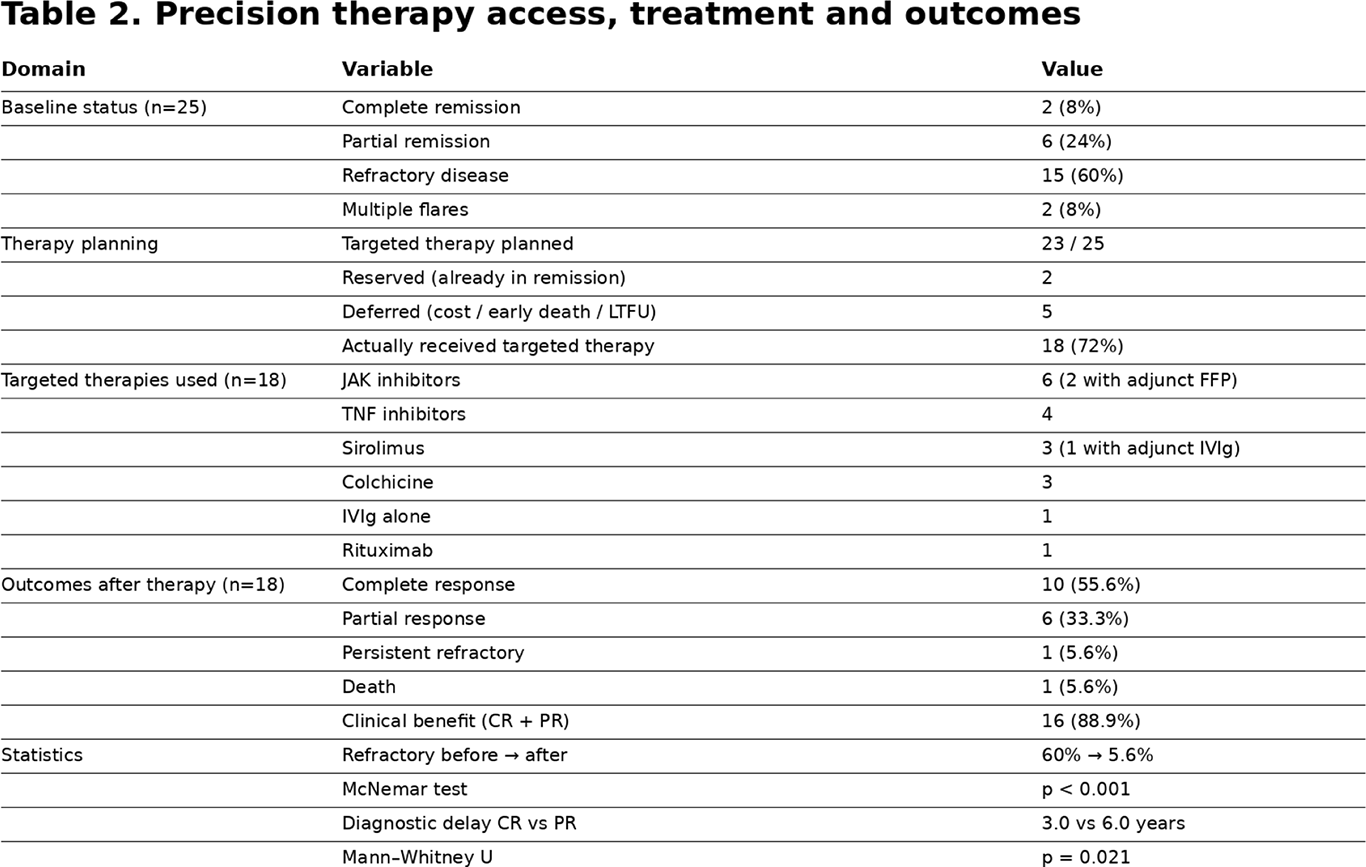
Before precision therapy, only 2/25 (8% ) were in complete remission and 6/25 (24% ) in partial remission, while 15/25 (60% ) had persistent refractory disease and 2 /25 (8% ) had multiple flares. Targeted therapy was planned in 23/25 , kept reserved in 2 who were already in remission, and deferred in 5 due to cost, early death or loss to follow-up, resulting in 18/25 (72% ) actually receiving pathway-aligned treatment. Initiated therapies included JAK inhibitors in 6 (with adjunct FFP in 2), TNF inhibitors in 4 , sirolimus in 3 (one with adjunct IVIg), colchicine in 3, IVIg alone in 1 and rituximab in one patient.
Among treated patients ( n=18 ), 10 (55.6%) achieved complete response , 6 (33.3%) partial response , 1 (5.6%) remained refractory and 1 (5.6%) died , yielding overall clinical benefit (CR+PR) in 16/18 (88.9% ). Within-patient comparison demonstrated a highly significant shift from 60% refractory disease before therapy to 5.6% after delivery of precision therapy ( McNemar test, p<0.001 ). Diagnostic delay was significantly associated with depth of response: partial responders had longer delay than complete responders ( median 6.0 vs 3.0 years; Mann–Whitney U, p=0.021 ).
Conclusions: This real-world study shows that genotype-guided, pathway-aligned therapy can fundamentally change the lives of children with severe immune dysregulation, transforming a largely refractory and steroid-dependent disease course into sustained clinical control for most patients. Children who received an earlier genetic diagnosis had better responses, proving that time to diagnosis is as important as the diagnosis itself. In resource-limited settings, the worst outcomes, including deaths and treatment failure, were seen where access to targeted therapy was delayed or not possible, highlighting that the main barrier is no longer medical knowledge but access to care. Precision immunogenetics should therefore not be viewed as an advanced option, but as an essential standard of care for every child with severe, atypical or treatment-resistant immune-mediated disease.
Table 1 .
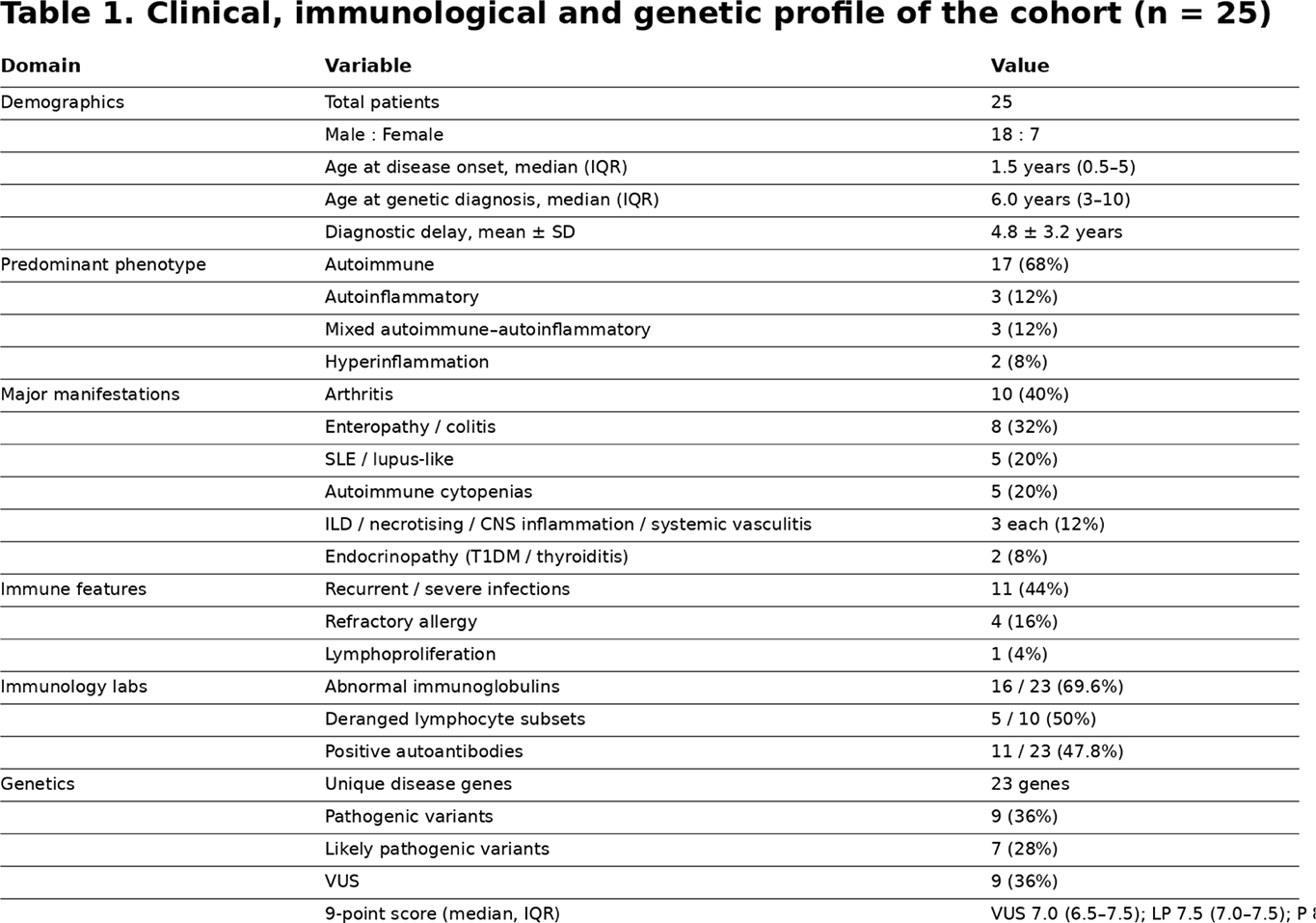
Table 2 .
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.