fetching data ... 
Background: We have previously developed and clinically validated an RNA-Seq based Molecular Signature Response Classifier (MSRC) to predict inadequate response to anti-TNF therapy in the treatment of Rheumatoid Arthritis. The MSRC is composed of 19 gene expression features along with anti- cyclic citrullinated peptide serostatus, sex, patient global assessment, and body mass index [1]. MSRC scores range from 1 to 25, with higher scores indicating a greater likelihood of inadequate TNFi response. Gene expression features are generated from total RNA sequencing (RNA-Seq) according to an analytically validated workflow [2]. In a clinical lab setting, multiple workflows are available for the extraction of RNA and generation of RNA libraries. Likewise multiple technologies for sequencing are available including commonly used platforms utilizing bridge amplification as well as more recent platforms which rely on rolling circle replication and DNA nanoballs).
Objectives: To demonstrate the robustness of the MSRC and its transcriptomic features, we performed analytical studies across two transcriptome sequencing workflows which utilize distinct RNA extraction and Library Preparation methods performed in different laboratories. In addition, we also measured performance using sequencing platform utilizing bridge amplification and compared the results with a platform using rolling circle amplification and DNA nanoballs. Ensuring high concordance across various platforms and workflows expands the accessibility of the test and increases confidence in the results across sequencing platforms and clinical laboratories.
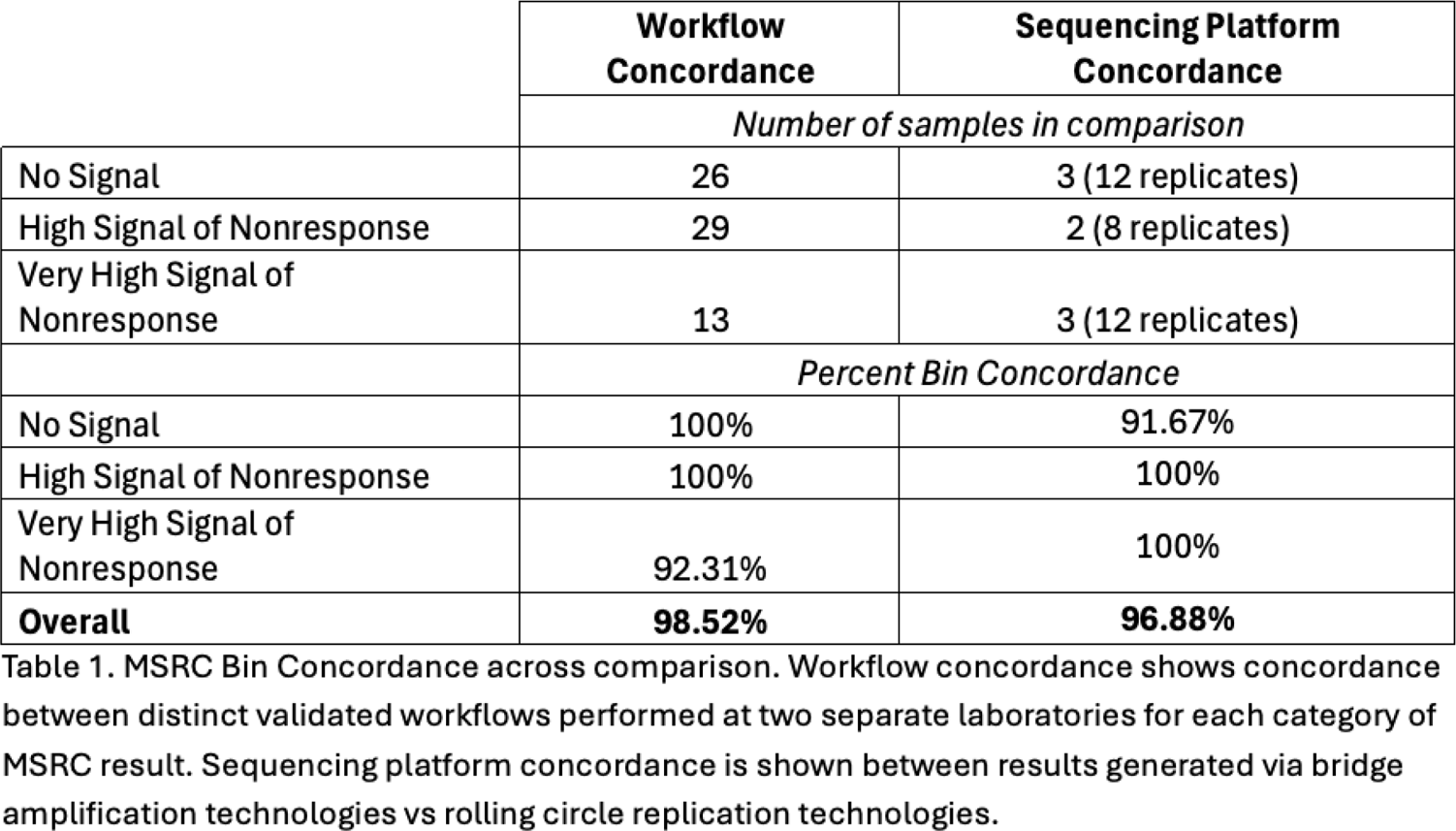
Methods: Two separate studies were performed. In one study, RNA extraction and library preparation workflows were compared. Whole blood was collected from each patient (n = 68 patients) and split between two laboratories where RNA was extracted, RNA libraries were generated in each laboratory via a distinct extraction and library preparation workflow. Sequencing was then performed via bridge amplification and results generated from samples processed at both laboratories were compared. In a separate study sequencing platforms were compared. RNA was extracted from the clinically validated workflow (n = 8 patients each with 4 technical replicates) and RNA libraries were generated using rolling circle replication and DNA nanoballs after which libraries were sequenced and results were compared with prior results generated using the standard validated workflow sequenced via bridge amplification. To assess concordance of results in each study, gene expression was quantified and regression analysis was performed and R 2 value was used to compare results from same samples prepared and sequenced in different laboratories. MSRC scores were bined by likelihood of nonresponse as No Signal (MSRC ≤ 10.6), High Signal of Nonresponse (MSRC > 10.6 and < 18.5), or Very High Signal of Nonresponse (MSRC ≥ 18.5), and concordance of bin assignment was assessed for each sample.
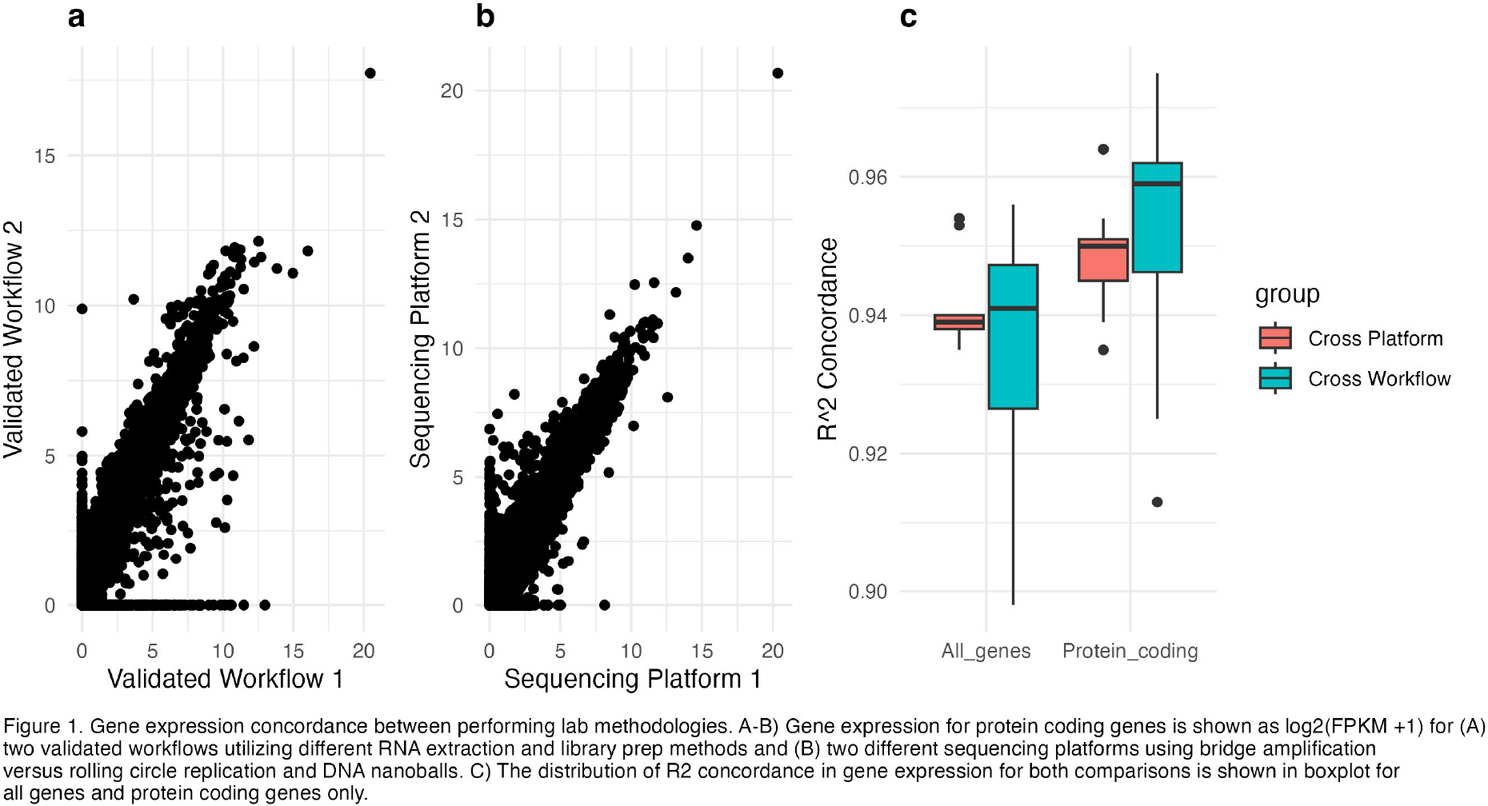
Results: Data was collected from patient samples sequenced through one of two validated workflows. Gene expression was quantified and expression was compared across the two workflows (Figure 1a). Concordance, as measured by R 2 value, had a median value of 0.94 across all genes and a median value of 0.96 for protein coding genes. MSRC bin concordance was 98.52% overall between workflows and only below 100% concordance among the Very High Signal of Nonresponse group (92.31%, Table 1). Separately we collected data from RNA which had alternately been sequenced using bridge amplification (platform 1) vs rolling circle replication (platform 2; Figure 1b). Gene expression concordance between platforms was high across all genes (median R 2 values = 0.939) and slightly better among protein coding genes only (median R 2 values = 0.95). Additionally, no significant difference was seen in the distribution of gene expression concordance for all genes or protein coding genes between cross workflow and cross platform comparisons (Figure 1C). MSRC Bin concordance between platforms was 96.88% overall and only below 100% in the No Signal group (91.67%; Table 1) indicating that the MSRC algorithm is robust to methodological differences in laboratory workflow and even across sequencing platforms.
Conclusions: In this study, we show evidence that the MSRC has high concordance and low technical variability across multiple workflows and sequencing platforms. These results demonstrate that the MSRC is agnostic to variability introduced by performing lab methodologies including differences in RNA extraction and library preparation methods, as well as to sequencing based on the commonly used bridge amplification technology or the more recent rolling circle amplification and DNAball technology. Together these data give confidence that the MSRC results will be consistent across multiple performing laboratories.
REFERENCES: [1] Cohen S et al. Rheumatol Ther. 2021 Sep;8(3):1159-1176.
[2] Jones A et al. Expert Rev Mol Diagn. 2021 Nov;21(11):1235-1243.
Acknowledgments: NIL.
Disclosure of Interests: None declared.