fetching data ... 
Background: Systemic sclerosis (SSc) is a severe autoimmune disease with limited therapeutic options. B cells and plasma cells play a central pathogenic role, and early clinical and tissue-level data suggest that BCMA×CD3 bispecific antibody therapy has the potential to enable targeted depletion of these pathogenic cell populations [1,2,3].
Objectives: To assess the efficacy and safety of the BCMA×CD3 bispecific antibody cizutamig in patients with severe diffuse cutaneous SSc, including clinical outcomes as well as molecular and cellular effects in blood, bone marrow, and skin.
Methods: Two patients with severe, progressive diffuse cutaneous SSc refractory to at least two prior immunosuppressive therapies were treated with cizutamig as individual treatment attempts after written informed consent. Treatment was administered intravenously with a single cycle of cizutamig (5 weekly doses). All immunosuppressive drugs were discontinued at least two weeks before treatment initiation. Due to the severe and progressive disease course, a second treatment cycle with an intensified dosing regimen was administered to Patient 2. Safety and efficacy were assessed over 29-32 weeks of follow-up.
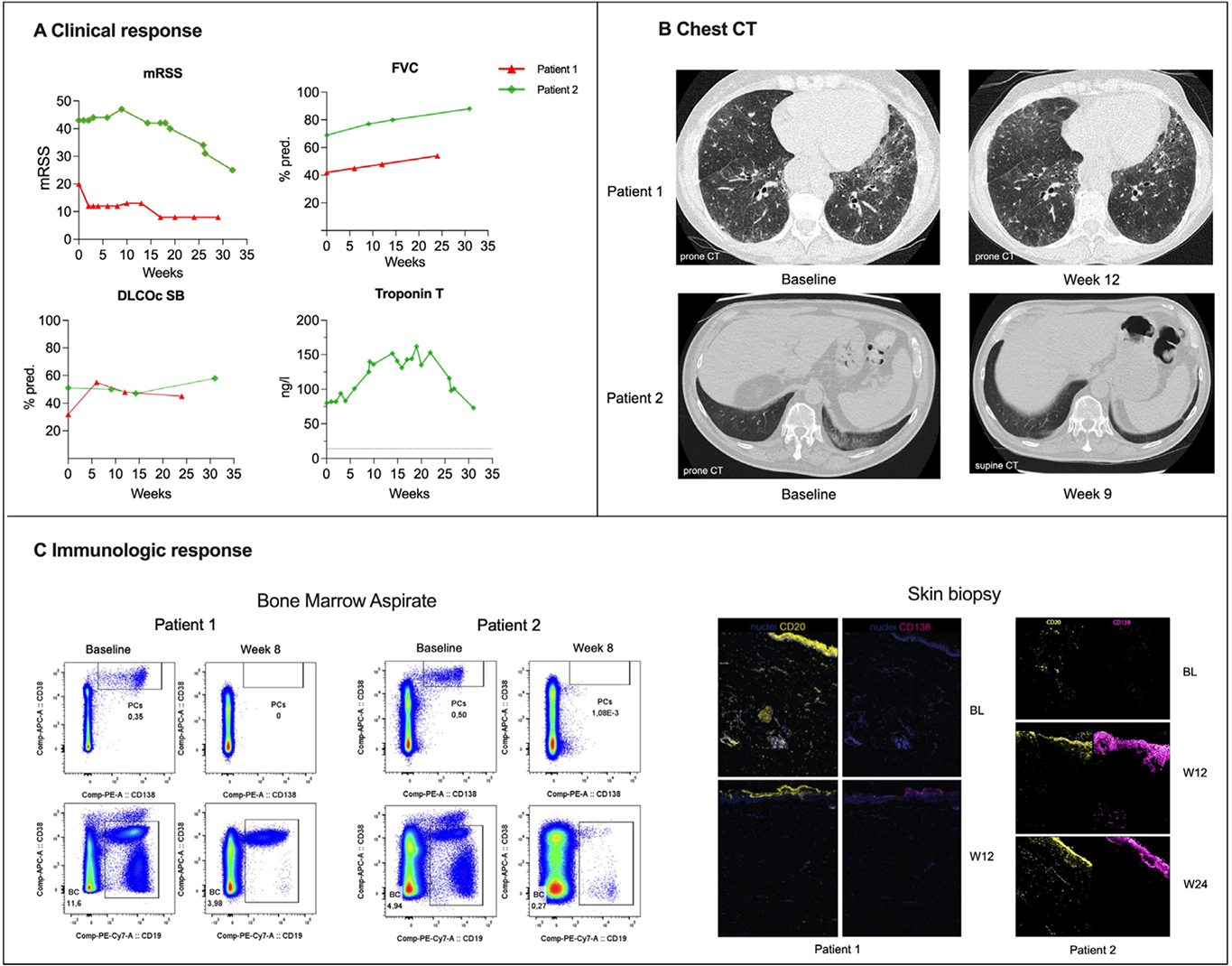
Results: Two patients with severe diffuse cutaneous SSc (anti-topoisomerase I-positive and anti-RNA polymerase III-positive) were treated with cizutamig. Baseline organ involvement included severe interstitial lung disease (ILD) in Patient 1 and severe skin involvement, cardiac involvement with mild ILD in Patient 2 (Table 1). There was no evidence of underlying malignancy in either patient. Cizutamig was generally well tolerated. Low-grade cytokine release syndrome was observed (Grade 1 in Patient 1 following the first dose, and mild Grade 2 based on fever and transient (< 30 min) asymptomatic hypotension (decrease in systolic blood pressure (SBP) to the mid-90s with baseline SBP in 100s-130s) immediately responsive to intravenous fluids in Patient 2 after the first dose); the latter treated with dexamethasone (10mg) and tocilizumab (600mg). No neurotoxicity was observed for either patient. Both patients developed hypogammaglobulinaemia (<4 g/L) requiring intravenous immunoglobulin. Patient 1 required hospitalisation 7 weeks after treatment initiation for SARS-CoV-2 infection and bronchitis and recovered following antiviral and antibiotic therapy. Patient 1 showed rapid and clinically meaningful improvement, achieving a revised ACR-CRISS 50 response by week 6, with marked improvement in lung imaging, pulmonary function tests (absolute increase in ppFVC from 42% to 52% and ppDLCO (cSB) from 32% to 48%) and skin involvement (12-point reduction in mRSS; Figure 1A and B) through 6 months of follow-up. These clinical improvements were accompanied by profound depletion of B cells and plasma cells across compartments, with complete or near-complete elimination of CD38 + CD138 + plasma cells and CD20 + B cells in peripheral blood by week 2, in bone marrow by week 8 and in skin by week 12 (Figure 1C).
Patient 2 demonstrated comparable systemic depletion in blood and bone marrow. However, plasma cells persisted in the skin after the first cycle, and given the severe and progressive disease course in the heart and skin, a second, intensified treatment cycle was administered, resulting in plasma-cell depletion in skin tissue, subsequent improvement of skin disease (mRSS maximum 47 to minimum 25), and heart (reduction of troponin from maximum of 152 to 73 ng/l and of pro-BNP from maximum of 2307 to 476 ng/l), together with improved functional capacity (absolute increase in ppFVC from 69% to 88% and ppDLCO (cSB) from 51% to 58%) and an increase in the six-minute walk distance from 0 m to 250 m using a rollator. Both patients exhibited reductions in antinuclear antibody titres. In Patient 2, a decrease of anti-topoisomerase I antibody of 36% was observed. Anti-RNA polymerase III-3-155 antibodies in patient 1 seroconverted within 6 months, while anti-RNA polymerase III-3-11 antibodies remained highly positive. Both patients achieved sustained clinical improvement following treatment with cizutamig without the need for any additional immunosuppression.
Conclusions: BCMA×CD3 bispecific antibody therapy with cizutamig was feasible and generally well tolerated in refractory diffuse cutaneous SSc, inducing deep B- and plasma-cell depletion with signals of clinical improvement or stabilisation. Further studies are required to define durability, optimal use, and long-term safety.
Table 1. Baseline Characteristics and Safety.
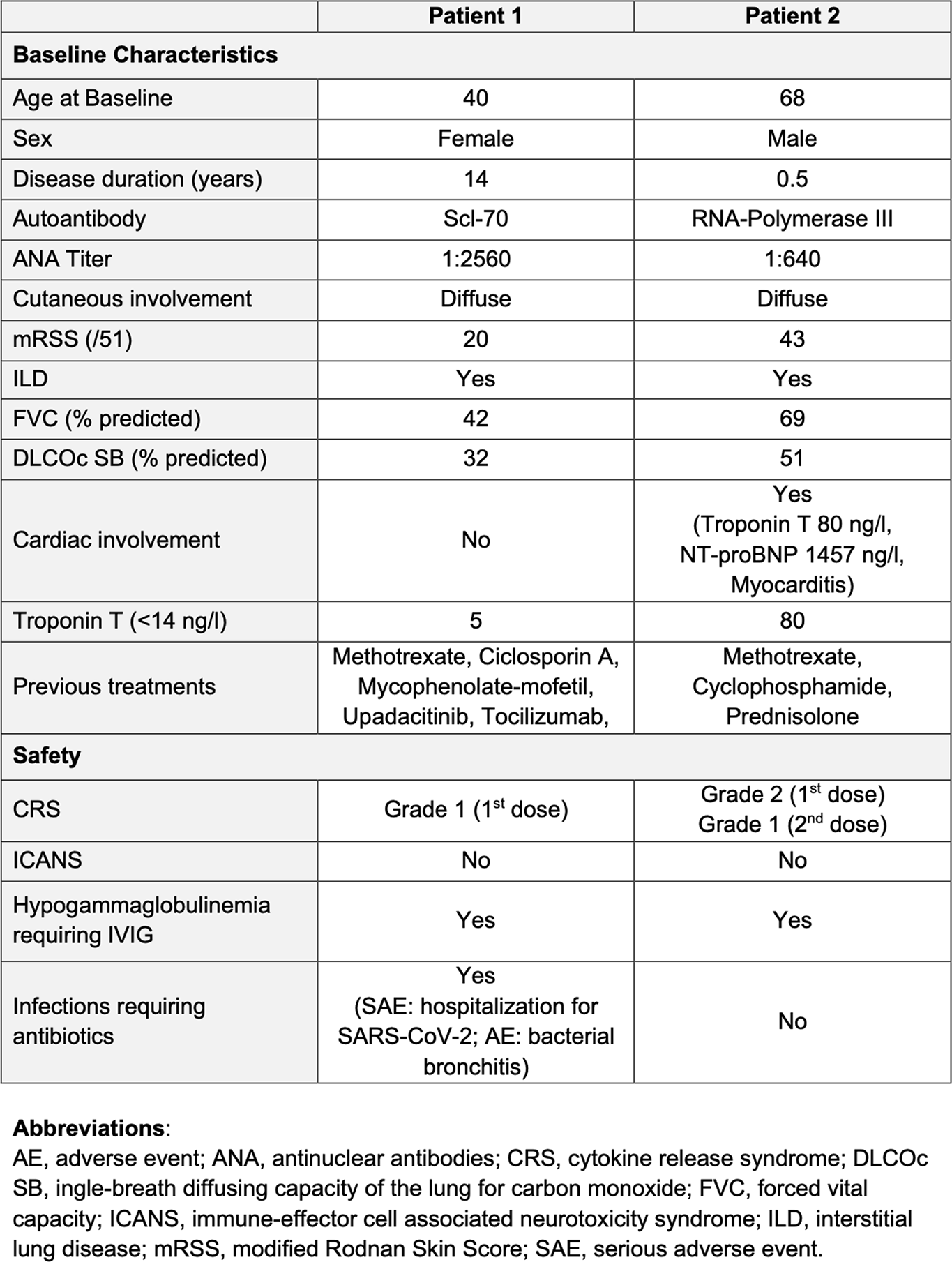
Clinical response, chest CT and immunologic response following treatment with cizutamig.
REFERENCES: [1] Scaletti C et al. Clin Exp Immunol. 2025.
[2] Siegert E et al. Ann Rheum Dis. 2025.
[3] Bucci L et al. N Engl J Med. 2025.
Acknowledgments: NIL.
Disclosure of Interests: Elpida Phithak: None declared, Robert Biesen: None declared, Elise Siegert: None declared, Fredrik N. Albach: None declared, Peter Wung Shareholder of Candid Therapeutics, Employee of Candid Therapeutics, Ken Song Shareholder of Candid Therapeutics, Employee of Candid Therapeutics, Tim Lu Shareholder of Candid Therapeutics, Employee of Candid Therapeutics, Jamie Haddon Shareholder of Candid Therapeutics, Employee of Candid Therapeutics, Philipp Krüger Shareholder of Candid Therapeutics, Employee of Candid Therapeutics, Arnd Kleyer: None declared, Maria Dzamukova: None declared, Arne Sattler: None declared, Marie Rehm: None declared, Edgar Wiebe: None declared, Anja Staeck: None declared, Qingyu Cheng: None declared, Stefano Bianco: None declared, Anja Fleischmann: None declared, Jan Zernicke: None declared, Tobias Alexander: None declared, Gerhard Krönke: None declared, David Simon: None declared.