fetching data ... 
Background: Systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by chronic immune activation, autoantibody production, and the formation of circulating immune complexes. These immune complexes deposit in target organs, particularly the kidney, where they activate complement and Fcγ receptors expressed by immune cells, leading to severe renal inflammation known as lupus nephritis (LN). LN develops in about 50% of patients within five years of diagnosis and represents a major determinant of morbidity and mortality, being associated with proteinuria, progressive loss of renal function, and increased cardiovascular risk. Fcγ receptors play a central role in immune complex driven inflammation. Among them, the transmembrane receptor FcγRIIA, expressed in humans but absent in mice, is a key activating receptor expressed by myeloid cells and platelets. We recently demonstrated that FcγRIIA-expressing platelets are required for firm neutrophil adhesion to immune complexes [1]. Consistently, in an FcγRIIA-humanized NZBxNZWF1 lupus model, platelet FcγRIIA exacerbated disease severity and accelerated lupus nephritis by promoting renal inflammation [2,3]. However, the downstream mechanisms regulating FcγRIIA dependent recruitment of inflammatory cells remain incompletely understood. Among factors that could modulate FcγRIIA-dependent responses, the 12-lipoxygenase (encoded by the ALOX12 gene) is a lipid-metabolizing enzyme highly expressed in platelets and certain myeloid populations and is responsible for the generation of bioactive lipid mediators, including the 12-hydroxyeicosatetraenoic acid (12-HETE). These mediators are closely linked to platelet activation and inflammatory signalling and have context-dependent pro-inflammatory or regulatory effects on leukocyte recruitment. Its contribution to FcγRIIA-dependent renal inflammation in lupus nephritis remains unknown.
Objectives: We hypothesize that 12-lipoxygenase and its lipid metabolites may regulate FcγRIIA dependent recruitment of leukocytes and platelets in lupus nephritis. Our objective is to determine the role of 12-lipoxygenase in lupus nephritis.
Methods: Renal lipidomic analyses were first performed in spontaneous NZBxNZW F1 lupus mice at different disease stages using mass spectrometry to profile 12-lipoxygenase–derived lipid mediators, including 12-HETE. To test the functional role of 12-lipoxygenase, FcγRIIA humanized C57Bl6/J mice ( FCGR2A TGN ) with or without the Alox12 gene ( FCGR2A TGN :: Alox12 +/+ vs FCGR2A TGN :: Alox12 −/− ) were used. Lupus nephritis was induced via injection of antibodies derived from sheep immunized with rat glomerular basement membrane (GBM), which deposit in the kidneys. Injection of these antibodies triggers the production of anti-sheep IgG, forming immune complexes in the glomeruli and mimicking renal inflammation. During these experiments, urinary albumin, a marker of renal injury, was measured. Kidneys were collected for immunofluorescence to quantify recruited neutrophils and platelets, and for lipidomic analyses using mass spectrometry. Circulating levels of 12-HETE were also measured in samples from patients with SLE and healthy individuals (n=70), and the correlation with platelet activation was determined by the evaluation of platelet activation markers in blood.
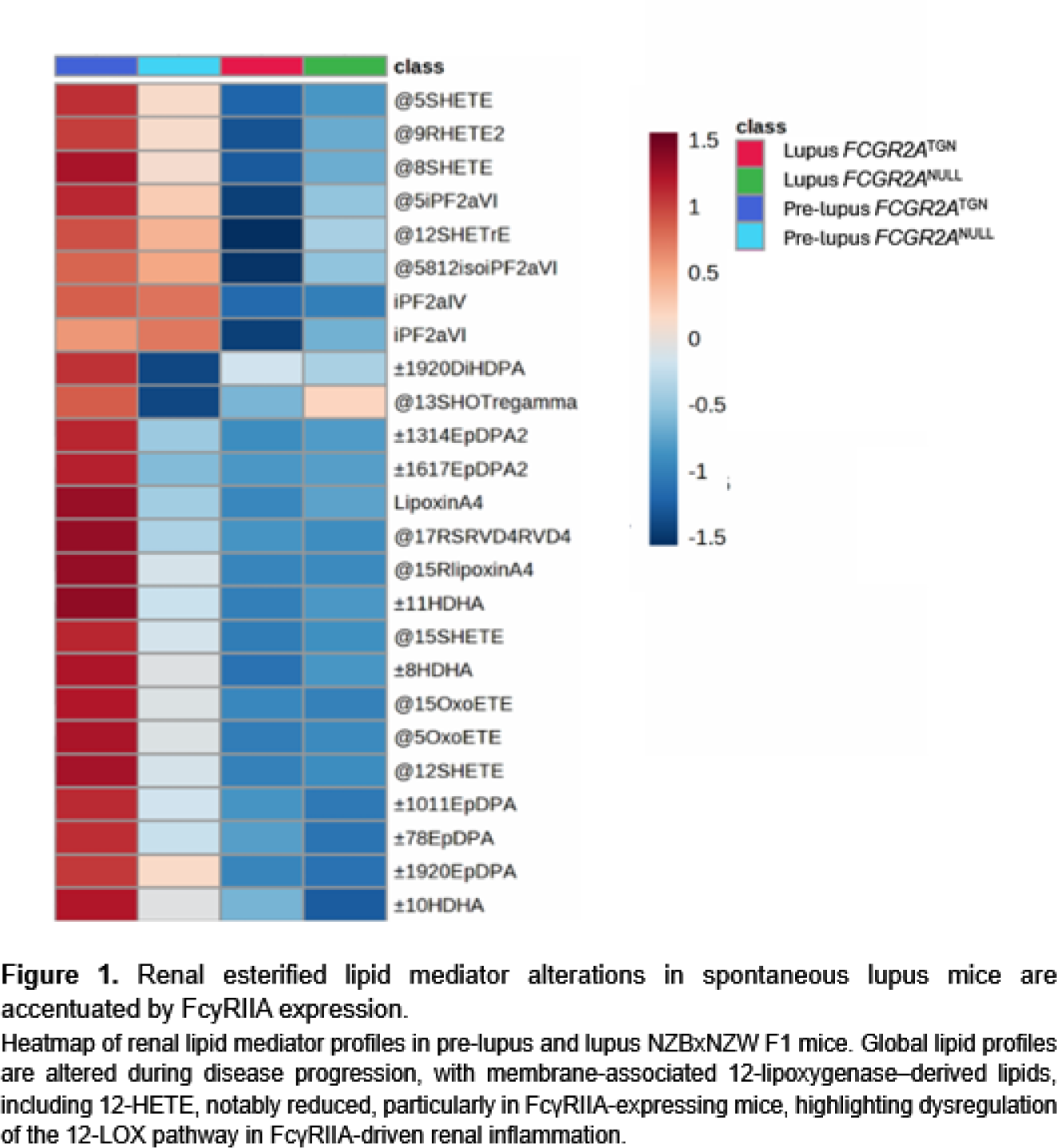
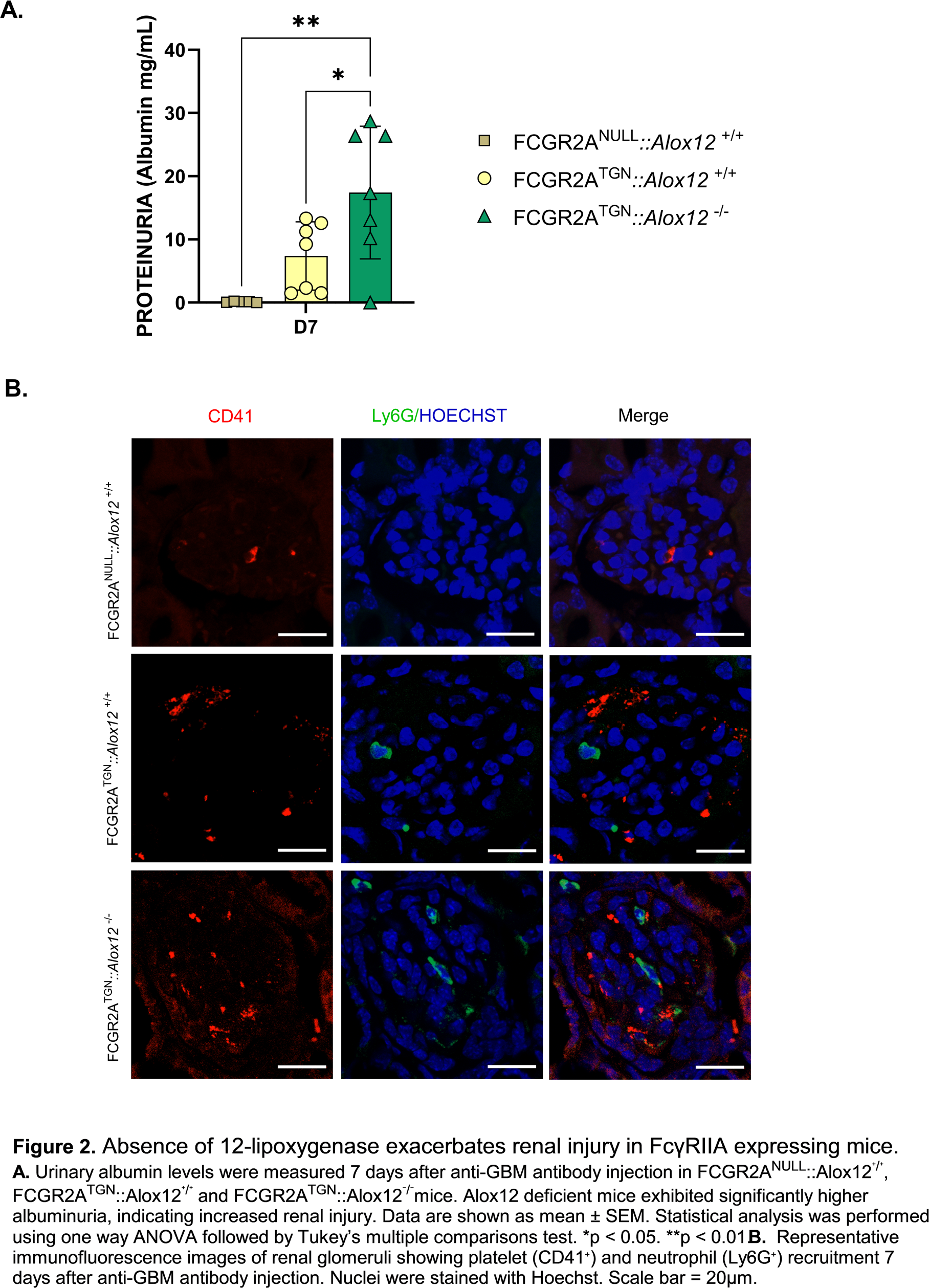
Results: Renal lipidomic analyses performed in the spontaneous NZBxNZWF1 lupus model revealed marked alterations in renal lipid mediator profiles during disease progression. Notably, membrane-associated (esterified) 12-lipoxygenase–derived lipids, including 12-HETE, were significantly reduced in kidneys from lupus mice compared with pre-lupus controls (Figure 1). This reduction was more pronounced in FcγRIIA-expressing mice, highlighting a dysregulation of the 12-lipoxygenase pathway in the context of FcγRIIA-driven renal inflammation. In the anti-GBM model seven days after anti-GBM antibody injection, FCGR2A TGN mice developed more severe nephritis than mice lacking FcγRIIA, as indicated by elevated proteinuria and increased complement deposition in the kidneys. The ablation of the Alox12 gene in FCGR2A TGN mice induced an aggravated proteinuria compared with FCGR2A TGN :: Alox12 +/+ mice, indicating aggravated renal injury (Figure 2.A). FcγRIIA expression promoted renal recruitment of neutrophils and platelets, and this effect was further enhanced in the absence of 12-lipoxygenase (Figure 2.B). Recruited cells were predominantly localized within the renal cortex, in the glomeruli. Consistently, circulating levels of 12-HETE were increased in patients with SLE and strongly correlated with platelet activation markers, supporting the relevance of 12-lipoxygenase pathway alterations in human disease involving platelets.
Conclusions: Renal leukocyte and platelet infiltration in lupus nephritis requires FcγRIIA, while the 12-lipoxygenase exerts a protective role by limiting nephritis and the renal recruitment of neutrophils and platelets. Dysregulation of 12-lipoxygenase derived lipid mediators, observed in both murine models and patients with SLE, suggests that this pathway contributes to the regulation of FcγRIIA dependent nephritis. Ongoing research aims to identify 12-lipoxygenase-derived mediators that may regulate inflammation. Understanding the regulatory role of these lipid mediators may guide the development of targeted therapeutic strategies to limit lupus nephritis in SLE patients.
REFERENCES: [1] Bellio M, Allaeys I, Doré E, Vaillancourt M, Lévesque T, Monteil M, et al. Immobilized IgG-containing immune complexes require platelets to recruit neutrophils during inflammation. J Clin Invest . In press.
[2] Melki I, Allaeys I, Tessandier N, Mailhot B, Cloutier N, Campbell RA, et al. FcγRIIA expression accelerates nephritis and increases platelet activation in systemic lupus erythematosus. Blood. 2020 Dec 17;136(25):2933–45.
[3] Melki I, Allaeys I, Tessandier N, Lévesque T, Cloutier N, Laroche A, et al. Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci Transl Med. 2021 Feb 17;13(581):5928.
Acknowledgments: NIL.
Disclosure of Interests: None declared.