fetching data ... 
Background: Anti-nuclear autoantibodies (ANAs) are detectable years before SLE classification. As preclinical autoimmunity progresses, some individuals develop limited SLE manifestations without meeting classification criteria (incomplete lupus erythematosus; ILE), forming a continuum from autoantibody-negative (aAb-) healthy individuals, through autoantibody-positive (aAb+) states and ILE, to established SLE. However, most aAb+ and ILE patients do not progress to SLE, indicating that additional, undefined immune changes drive transition to clinical disease.
Objectives: To define cell type- and pathway-specific immune alterations across the evolution of pre-SLE states and to identify immune signatures associated with the transition from preclinical autoimmunity to clinical disease.
Methods: aAb- healthy controls (n=12), aAb+ healthy individuals (n=15), ILE patients (n=15), and SLE patients (n=18) were classified by clinical criteria and SLE-associated autoantibodies measured by Bioplex 2200 and indirect immunofluorescence. aAb+ individuals had > 1 of 13 SLE-associated autoantibody specificities, and ILE patients had > 1 autoantibody and ACR SLE classification scores of 2-3 without meeting full criteria. Transcriptomes and surface proteins were profiled using 5’ single-cell immune profiling with 137-plex CITE-seq. Disease-stage signatures were identified using logistic regression, gene set enrichment, and machine learning models. Genetic ancestry was inferred from Infinium Global Screening Array data and included, along with age, as covariates in all models. Cell communication analysis was performed with the Liana Python package. Over 5,400 serum proteins were quantified in 67 subjects (ANA-, ANA+ healthy, ILE, SLE) using Proximity Extension Assay (Olink Explore HT).
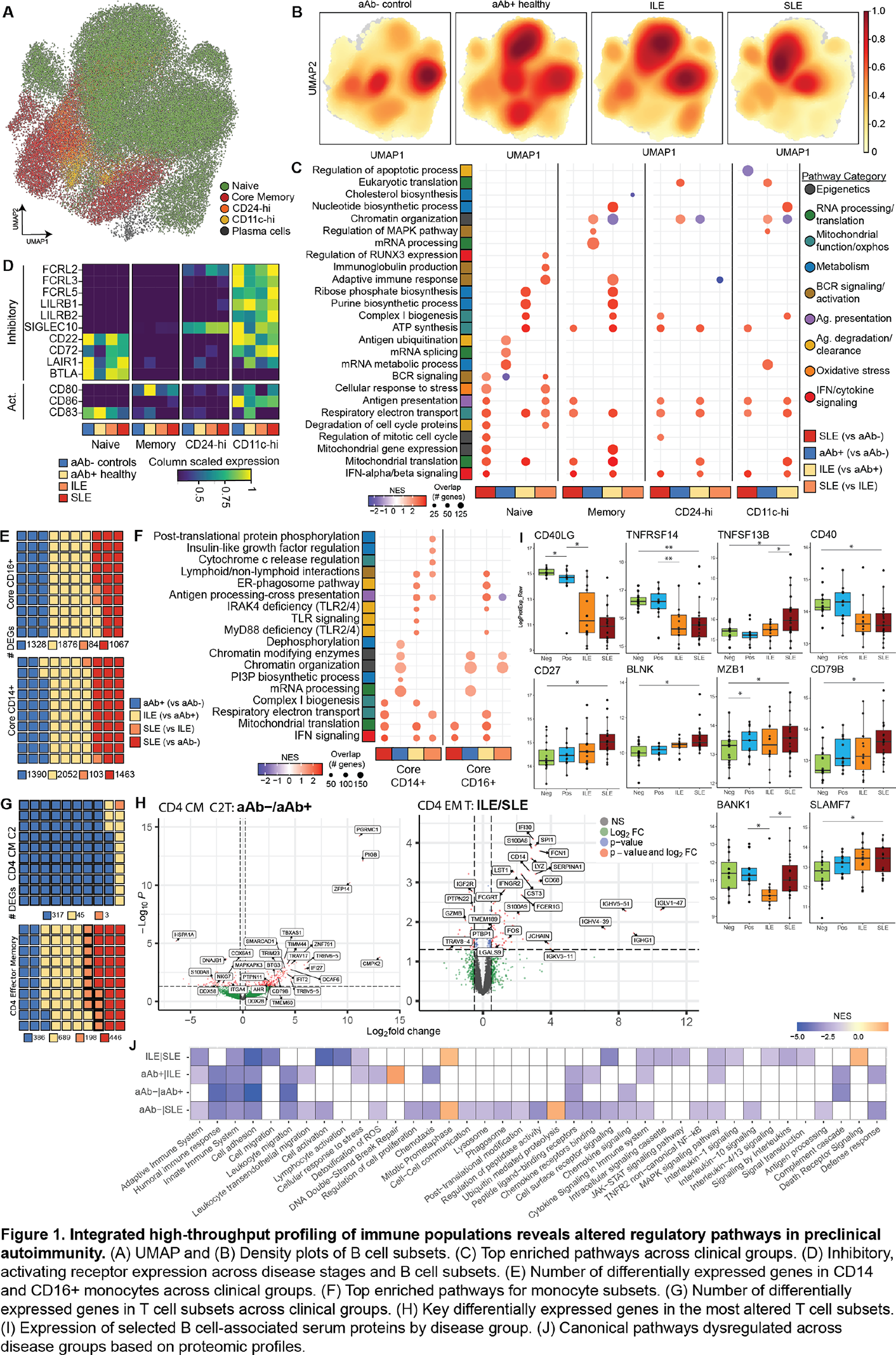
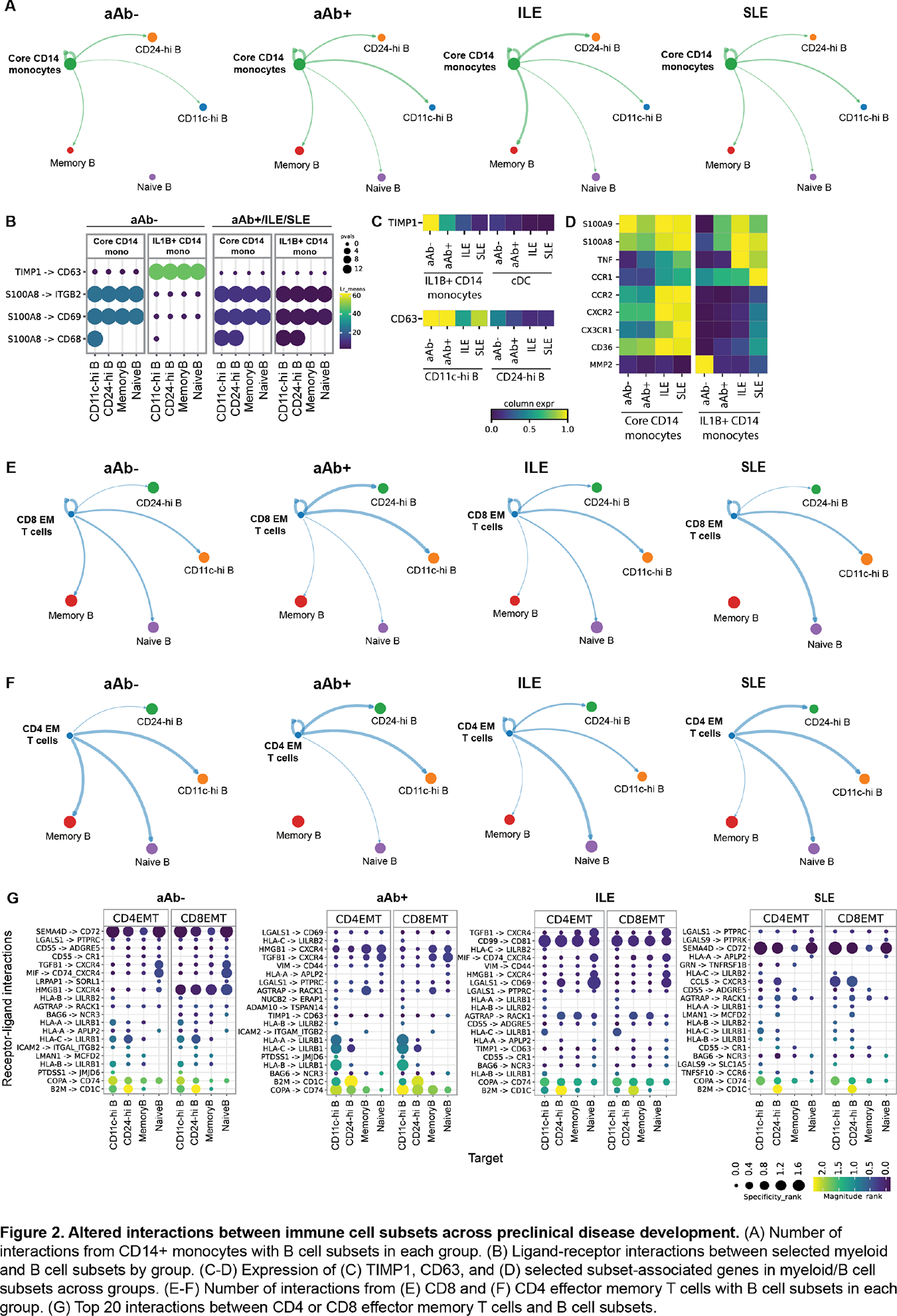
Results: In B cells, density projections showed disease-associated shifts in naïve, memory CD24-hi, and CD11c-hi subsets across the aAb- to SLE continuum (Figure 1A-B). Pathway analyses showed convergence of all B cell subsets on inflammatory programs of SLE, including type I IFN signaling, antigen presentation, and mitochondrial oxidative metabolism (Figure 1C). Antigen-experienced subsets (memory, CD24-hi, CD11c-hi) acquired biosynthetic, type I IFN, and mitochondrial signatures early at the aAb+ or ILE stages (Figure 1C). In contrast, naïve B cells followed a slower trajectory, with metabolic reprogramming emerging in ILE, and full BCR activation and effector signatures appearing predominantly in SLE (Figure 1C). Surface-receptor profiles showed loss of naïve B cell tolerance checkpoints at the aAb+ stage, with downregulation of inhibitory receptors (CD22, CD72, LAIR1, BTLA) and induction of the activation marker CD83, while memory B cells upregulated the costimulatory receptor CD80 and CD11c-hi cells retained high expression of FCRL and SIGLEC10 across the continuum (Figure 1D), indicating subset-specific remodeling of activation and regulatory circuits. In monocytes, CD14+ and CD16+ subsets showed the largest transcriptional changes, particularly between aAb+ and ILE (Figure 1E). Specifically, chromatin-organization and mRNA-processing pathways increased in aAb+ individuals, and a broad induction of type I IFN, mitochondrial, inflammasome, chemotaxis, and cytokine-production programs in ILE and SLE (Figure 1F), consistent with progressive priming toward a hyper-inflammatory state. Among T cells, differential expression in aAb+ versus aAb- was enriched in CD4 central memory T cells, whereas differences between ILE and SLE localized to CD4 effector memory T cells (Figure 1G-H). Serum proteomics demonstrated stage-dependent remodeling of B cell-associated proteins and broad dysregulation of adaptive and innate immune, trafficking, and stress-response pathways along the aAb- to SLE continuum (Figure 1I-J). Ligand-receptor analysis revealed loss of TIMP1-CD63 interactions from IL1B+ CD14+ monocytes to multiple B cell subsets, driven by progressive downregulation of TIMP1 in IL1B+ CD14+ monocytes and reduced CD63 expression on CD24-hi and CD11c-hi B cells. In parallel, S100A9-driven signaling toward B cell receptors increased along the disease continuum, accompanied by higher expression of S100A9 in IL1B+ CD14+ monocytes together with elevated TNF, IL6, and other inflammatory mediators (Figure 2A-D), suggesting that reprogrammed monocytes shift from providing pro-survival regulatory signals to delivering inflammatory cues that support autoreactive B cell activation. In T cells, most differences in B-T cell communication involved CD8 and CD4 effector memory T cells, which gained multiple interactions with naïve, memory, CD24-hi, and CD11c-hi B cell subsets across the disease continuum (Figure 2E-G).
Conclusions: Preclinical autoimmunity is characterized by early disruption of naïve B cell tolerance checkpoints, progressive inflammatory reprogramming of monocytes, and increased engagement of effector memory T cells with B cell subsets. Rewired monocyte-B cell communication, including loss of TIMP1-CD63 signals and emergence of S100A9-driven inflammatory pathways, together with expanded CD4/CD8 effector memory T cell interactions with B cells, is associated with activation and remodeling of antigen-experienced B cells in clinical SLE. These findings identify specific B cell-myeloid-T cell interactions as testable early intervention targets to prevent progression from silent autoimmunity to SLE.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: Aleksandra Bylinska: None declared, Miles Smith: None declared, Rufei Lu: None declared, Catriona Wagner: None declared, Ben Jones: None declared, Matthew Caleb Marlin Standard Biotools Inc, Carla Guthridge: None declared, Christian Wright: None declared, Susan Macwana: None declared, Wade DeJager: None declared, Marci Beel: None declared, Christopher J Lessard Johnson and Johnson Innovative Medicine Research Alliance Sjögren’s Disease Advisory Board, research support from Johnson and Johnson Innovative Medicine (formerly Janssen; ended 12/31/2023), Cristina Arriens Advisory or review panels: Biogen, Bristol Myers Squibb, Cabaletta, Health & Wellness Partners, Synthekine, AstraZeneca (grant support for an Investigator-Initiated study), Teresa Aberle: None declared, Joan Merrill Taught SLE disease outcome measures for Biogen, AbbVie, Zenas, AbbVie, Amgen, AstraZeneca, Boehringer Ingelheim, BMS, EMD Serono, Genentech, Gilead, GSK, Lilly, Merck, Novartis, Pfizer, RemeGen, Sanofi, UCB and Zenas, Astra Zeneca, BMS, Joel M Guthridge: None declared, Judith A. James GSK, OMRF licensed IP to Progentec Biosciences.