fetching data ... 
Background: CD19 targeted autologous chimeric antigen receptor (CAR) T cells continue to demonstrate promising clinical activity across an array of B-cell driven autoimmune diseases. However, access in underserved regions has been limited as occurrences of safety outcomes associated with CAR T-cell treatment and the requirement of harsh conditioning chemotherapy (CCT) regimens associated with severe toxicities (e.g. cytopenias and severe opportunistic infections) necessitates prolonged hospitalization. Moreover, the need for patients to pause immunosuppressive treatments, logistical challenges in manufacturing that induce delays, product inconsistencies, and high cost, all represent obstacles to their widespread adoption in autoimmune disease. Furthermore, the immunopathogenesis of autoimmune diseases is often driven by complex and heterogeneous subsets of B, T, and NK cells that are incompletely targeted by existing single mechanism of action (e.g. CD19 or BCMA) CAR T-cell therapies.
Objectives: FT839 is an off-the-shelf, multi-point engineered dual-CAR T-cell product targeting both lineage and disease state cells, developed to overcome the single antigen targeting limitation of existing autologous T-cell products, eliminate the need for CCT, and expand therapeutic delivery, application, and safety to enable expanded reach in diverse autoimmune disease treatment settings.
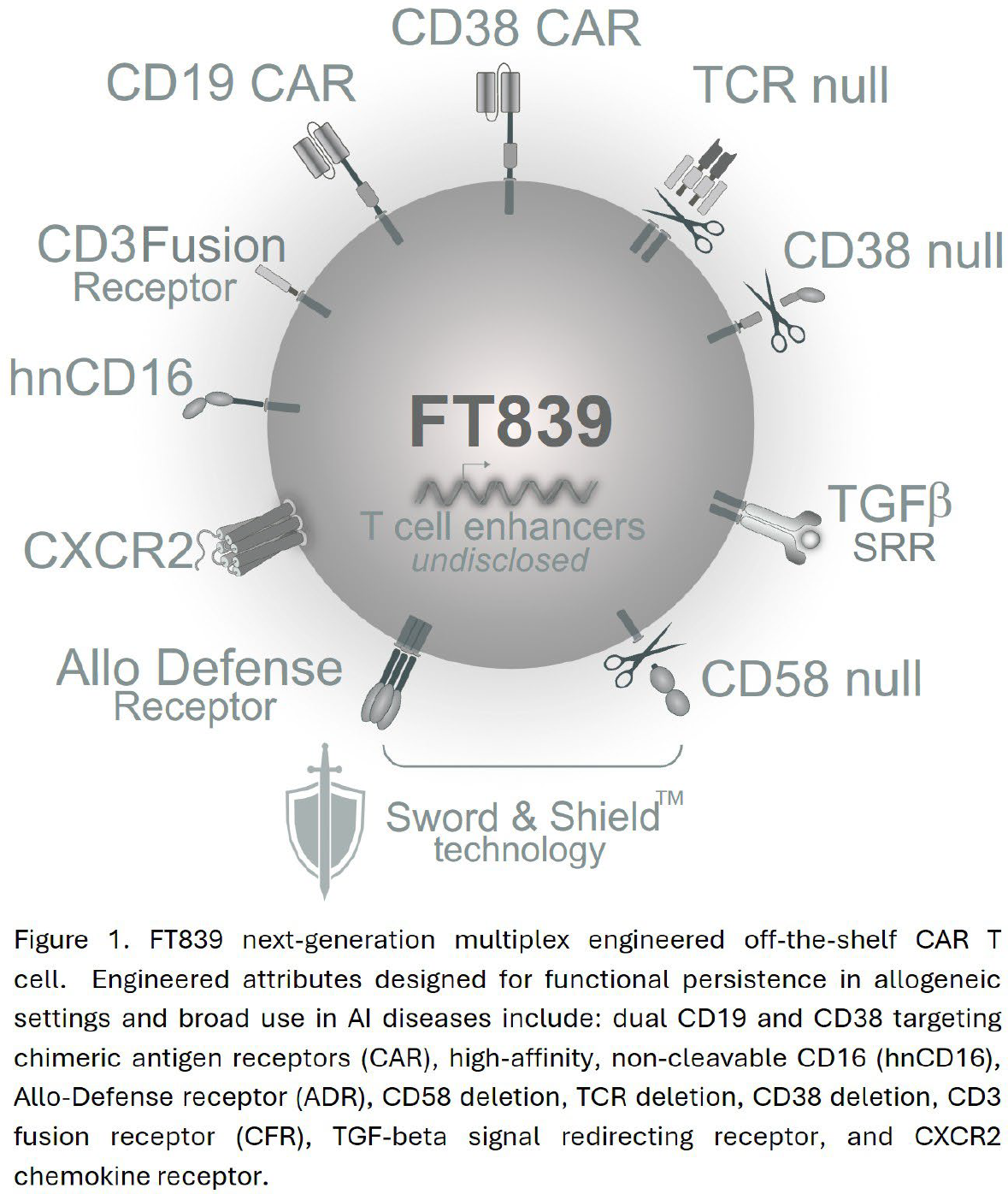
Methods: FT839 is derived from a clonal and multiplex engineered induced pluripotent stem cell (iPSC), enabling its standardized, consistent, cost-effective, and scaled manufacture. The engineering profile of FT839 incorporates novel synthetic elements that impart the cell product flexible multi-antigen targeting intended to eliminate a range of pathogenic immune cell types, and resistance to host-mediated allogeneic immune rejection (Figure 1). FT839 expresses two unique CARs targeting (i) the B-cell lineage marker CD19 and (ii) the immune activation marker CD38, respectively. FT839 is also engineered to express a high-affinity, non-cleavable Fc receptor (hnCD16) and a chimeric CD3-fusion receptor (CFR), extending the targeting capability and depth of function of FT839 in combination with standard-of-care monoclonal antibodies or clinically approved T cell engagers, respectively. To avoid host-mediated alloimmune rejection FT839 features dual Sword and Shield engineering, incorporating a synthetic alloimmune defense receptor (ADR) that selectively targets and eliminates 4-1BB + alloreactive immune cells and the genetic deletion of CD58 to limit host immune cell recognition.
Results: Single cell RNA sequencing from healthy donor peripheral blood mononuclear cells (PBMCs), demonstrated that expression of CD19 and CD38 define a broad set of B, NK, and activated T cell subsets [1] that mediate and maintain many autoimmune diseases, including: Rheumatoid Arthritis, Myasthenia Gravis, Type I Diabetes, Psoriatic Arthritis, Multiple Sclerosis, and Systemic Lupus Erythematosus, among others. In short-term cytotoxicity assays against PBMCs from healthy donors, supplemented with differentiated plasmablasts and plasma cells, FT839 selectively eliminated CD19+CD38+/- B cells (>99% CTL), CD19lowCD38+ plasmablasts (~99% CTL), CD19-CD38+ plasma cells (>99% CTL), and CD38+ activated CD4+ and CD8+ T cells (98.1% & 93% CTL, respectively). Importantly, no specific cytotoxicity by FT839 was observed against non-activated, CD38 negative T cells (0% CD38-CD4+ CTL; 0% CD38-CD8+ CTL). FT839 similarly demonstrated potent and specific elimination of leukemia and lymphoma target cells that differentially express CD19 and/or CD38. Depth of cytotoxicity was further improved when combined with a CD20 specific mAb to activate hnCD16 or a CD20 specific T cell engager to activate CFR. The ability of FT839 to avoid host-mediated alloimmune rejection was demonstrated in extended mixed-lymphocyte reaction cultures with healthy and diseased HLA-mismatched PBMCs. Through Sword and Shield engineering, FT839 specifically suppressed the emergence of an alloreactive and activated CD25 + 4-1BB+ T cell population while selectively eliminating both CD19 and CD38 expressing immune cell subsets. Non-activated/non-alloreactive T cells were preserved vis-a-vis enhanced persistence of FT839 (2.4e6 vs 1.3e4; FT839 vs no Sword/Shield; p<0.0001). In the presence of alloreactive PBMCs, FT839 demonstrated sustained CD19 and CD38 specific cytotoxicity upon serial rechallenge in vitro and maintained tumor growth inhibition and functional persistence in vivo, consistently displaying superior durability and potency compared to control cells that lacked Sword and Shield engineering. These results demonstrate the unique ability of FT839 to evade host-mediated alloimmune rejection, achieve functional persistence in allogeneic and mismatched settings and obviate the need for CCT.
Conclusions: FT839 has durable and persistent function in allogeneic settings where it selectively and flexibly eliminates multiple disease-driving immune cells without the need for toxic conditioning chemotherapy. These capabilities are further enhanced in combination with clinically approved mAbs or T cell engagers. Its scalable, cost-effective, and consistent manufacturing process and off-the-shelf delivery supports broad clinical accessibility, thereby extending applicability across a range of diverse B and T-cell driven autoimmune diseases.
REFERENCES: [1] Gong Q, Sharma M, Glass MC, et al. Multi-omic profiling reveals age-related immune dynamics in healthy adults. Nature . 2025;648(8094):696-706.
Acknowledgments: NIL.
Disclosure of Interests: Natalie J. Shiff Shareholder: Fate Therapeutics, Inc., Gilead, CRISPR, Roche, Novovax, Cabaletta, AbbVie, Vertex., Employed by Fate Therapeutics, Inc., Mark Jelcic Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Shilpi Chandra Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Soheila Shirinbak Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Brian Groff Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Alan Williams Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Alma Gutierrez Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Miguel Meza Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Bjoern Gaertner Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Karina Palomares Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Loraine Campanati Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Carissa Dege Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Pei-Fang Tsai Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Tom Lee Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Raedun Clarke Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Karl-Johan Malmberg Research support: Fate Therapeutics, Inc., Maksim Mamonkin Research support: Fate Therapeutics, Inc., Bahram Valamehr Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Vaneet Sandhu Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Alex Garcia Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Jode Goodridge Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc., Martin Hosking Shareholder: Fate Therapeutics, Inc., Employed by Fate Therapeutics, Inc.