fetching data ... 
Background: Sjögren’s disease (SjD) is a female-dominant, systemic autoimmune rheumatic disease with high unmet medical need. Patients with SjD are generally beset by pain, fatigue, and dryness and 30%–50% develop heterogeneous systemic involvement. Heterogeneity of disease etiology and clinical presentation have resulted in significant challenges in developing effective therapeutics. Biomarker data from historical SjD trials could be used to inform future trials and increase probabilities of success through strategies such as identifying dysregulated pathways associated with clinical and pathological endotypes [1]. SjD can present with several other autoimmune diseases but has a particularly high concordance with systemic lupus erythematosus (SLE), as 23%–35% of patients with SLE develop associated secondary SjD [2]. The commonality between these two diseases is supported by mechanistic overlap involving type 1 interferons and underlying genetics, which have identified tyrosine kinase 2 (TYK2) and TYK2-related pathways conferring susceptibility [3]. Deucravacitinib is a selective TYK2 inhibitor (TYK2i) that has shown clinical efficacy in the phase 2 PAISLEY SLE trial 4 and with active phase 3 investigations in SLE and SjD.
Objectives: We aimed to investigate the transcriptomic link between SLE and SjD in the context of a single sample gene set enrichment analysis (ssGSEA) TYK2i signature and to establish the relationship between TYK2i signature and clinical parameters and disease domains of SjD. The TYK2i signature is a pattern of changes in gene activity that shows how the immune system responds when TYK2 is blocked by a drug.
Methods: Early transcriptomic pharmacodynamic (PD) responses to deucravacitinib were assessed in patients with SLE from the PAISLEY SLE trial (NCT03252587) and used to generate a TYK2i signature. In this phase 2, double-blind, placebo-controlled trial, patients with active SLE were randomized 1:1:1:1 to receive placebo, deucravacitinib 3 mg twice daily (BID), deucravacitinib 6 mg BID, or deucravacitinib 12 mg once daily (QD) for 48 weeks [4]. Bulk RNAseq was performed on whole blood from a subset of patients within 72 hours of initial dose of placebo (n=18) or deucravacitinib 3 mg BID (n=22). Significantly downregulated genes from baseline in the deucravacitinib 3 mg BID arm were identified and aggregated to create a ssGSEA TYK2i signature. Data from patients with SjD were obtained from a phase 3, double-blind, placebo-controlled trial (NCT02915159) in which patients with active SjD were randomized 1:1 to receive placebo or abatacept weekly for 169 days [5]. Whole blood was collected from patients (n=186) at baseline and used for bulk RNAseq to identify transcriptomic differences in SjD patients vs age- and sex-matched normal healthy volunteers (NHV; n=45). TYK2i signature was applied to baseline whole blood RNAseq from patients with SjD and compared with NHV by Kruskal-Wallis test. Spearman and Pearson correlations were performed between baseline TYK2i signature and baseline clinical disease severity of the overall EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI) and associated subdomains where distributions were of sufficient sample size for modeling. Additional correlation models were performed identifying relationships between baseline TYK2i signature and clinical laboratory measures. Baseline autoantibody levels were stratified by cutoff levels and compared with TYK2i signature levels using Wilcoxon rank-sum tests.
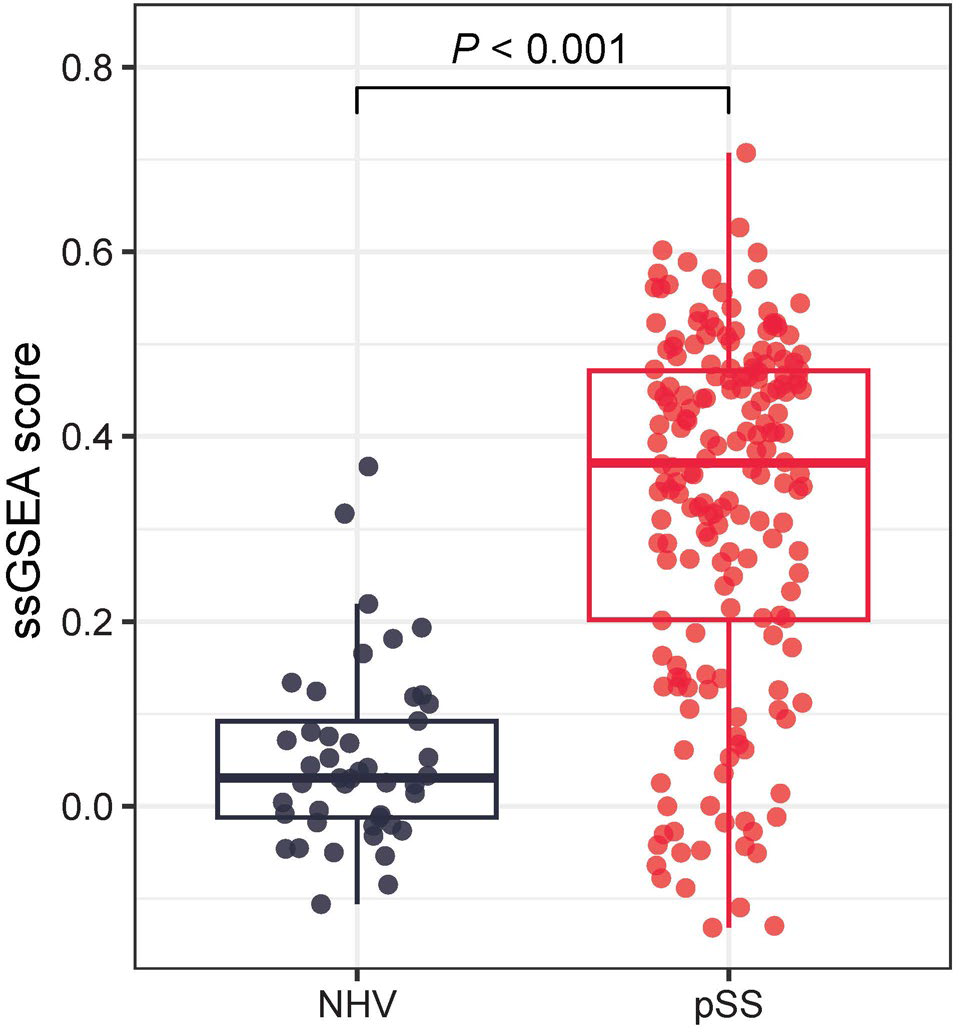
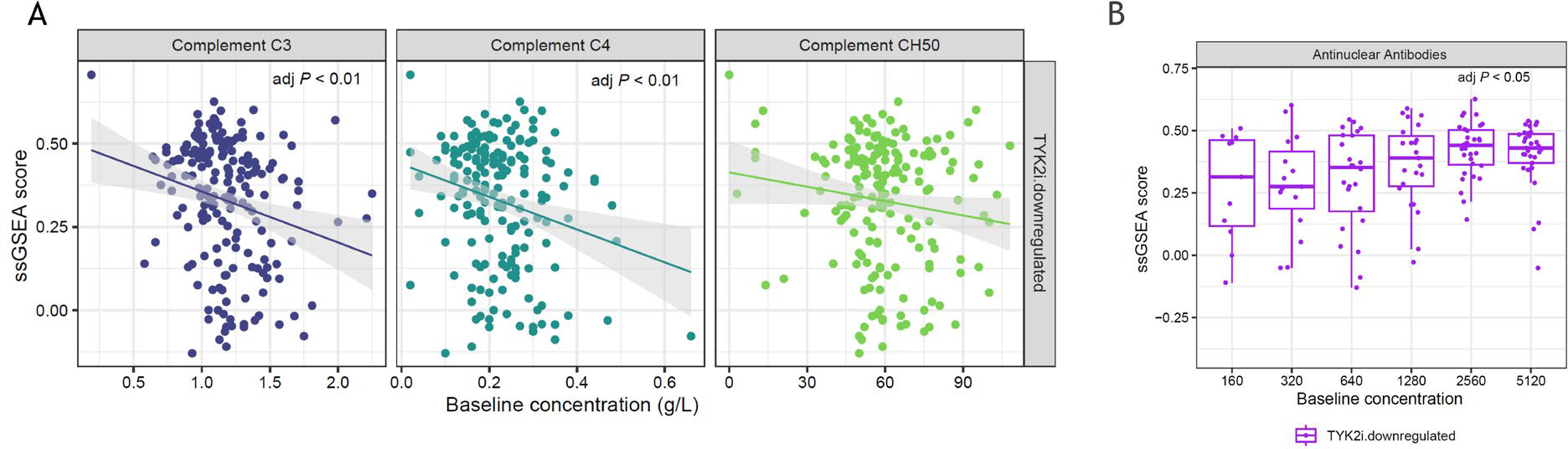
Results: Transcriptomic analysis of direct PD effects of deucravacitinib in patients with SLE revealed significantly downregulated expression of 75 genes within 72 hours of treatment initiation, after fold change (FC) multiplicity adjustment (FC ≥1.5, adj P <0.05), while no genes in patients on placebo reached this threshold. Of the 75 genes, 71 were detected in the SjD cohort and were used to create a SjD-relevant TYK2i signature. TYK2i signature at baseline in patients with SjD was significantly elevated relative to NHV ( P <0.001) (Figure 1). Total ESSDAI was not significantly correlated with TYK2i signature in patients with SjD; however, ESSDAI subdomains positively correlated with TYK2i signature, including biological (adj P <0.001) and lymphadenopathy (adj P <0.05). Baseline complements C3 and C4 (Figure 2A) and cell counts including leukocytes, basophils, and neutrophils negatively correlated with TYK2i signature. Total IgG (adj P <0.001) and IgA (adj P <0.05), but not IgM, were significantly correlated at baseline with TYK2i signature. TYK2i signature was significantly elevated in patients displaying higher concentrations of baseline anti–Sjögren’s-syndrome-related antigen A autoantibodies and anti-nuclear antibodies compared with lower concentrations (Figure 2B).
Conclusions: In a phase 2 SLE clinical trial, deucravacitinib induced a significant reduction in the expression of genes indicative of pathways directly regulated by TYK2 as early as 72 hours after drug initiation. This TYK2i gene signature was significantly upregulated at baseline in patients with SjD compared to NHV, supporting evidence of common dysregulated pathobiology between SLE and SjD. TYK2i signature correlated with baseline severity of SjD clinical symptoms and laboratory abnormalities. These data support robust biological evidence of TYK2 pathway dysregulation relevant to key clinical manifestations in SjD, providing strong rationale for the investigation of TYK2i deucravacitinib in the treatment of SjD.
Increase of TYK2i signature in patients with SjD vs NHV
Association of TYK2i signature with baseline (A) complement levels and (B) anti-ANA in patients with SjD
REFERENCES: [1] Tarn JR, et al. Lancet Rheumatol 2019;1:e85–e94.
[2] Gianordoli APE, et al. Adv Rheumatol 2023;63:11.
[3] Khatri B, et al. Nat Commun 2022;13:4287.
[4] Morand E, et al. Arthritis Rheumatol 2023;75:242–252.
[5] Baer AN, et al. Ann Rheum Dis 2021;80:339–348.
Acknowledgments: NIL.
Disclosure of Interests: Brandon Johnson Bristol Myers Squibb, Bristol Myers Squibb, Jacob Bumgarner I was employed by Bristol Myers Squibb, Shangzhong Li I worked in Pfizer before and were granted stock., I worked in Pfizer between 2019 to 2023 before joining BMS, Chun Wu Bristol Myers Squibb, Bristol Myers Squibb, Michele Bombardieri Amgen, Novartis, Amgen, Novartis, sGTX, ArgenX, GlaxoSmithKline, Bristol Myers Squibb, Elena Pontarini: None declared, Xavier Mariette Alfasigma, BMS, GSK, Jonsson, Novartis, Pfizer, Divi Cornec Novartis, GSK, BMS, Roche-Chugai, CSL Behring, Astra-Zeneca, Sanofi, Jinqi Liu Bristol Myers Squibb, Bristol Myers Squibb.