fetching data ... 
Background: The cause of chronic type I interferon (IFN-I) activation in autoimmune diseases such as Sjögren’s disease (SjD) remains unclear. Although viral infections have long been suspected, there is limited evidence of persistent viral infection in patient tissues. Increasing evidence suggests that epithelial cells, the main target cells in SjD, can themselves generate internal danger signals that drive ongoing immune activation. Double-stranded RNA (dsRNA) is a powerful trigger of IFN responses, yet how epithelial cells normally control endogenous dsRNA is poorly understood. RNA N6 -methyladenosine (m 6 A) modification is an epitranscriptomic regulatory mechanism that regulates RNA outcomes in cells and is controlled by the enzyme METTL3. While m 6 A has emerged as an important regulator of immune balance, its role in limiting epithelial-driven IFN activation in autoimmune disease remains unknown.
Objectives: To determine whether defective m 6 A dependent RNA regulation in epithelial cells leads to inappropriate IFN-I signature contributing to the initiation and maintenance of SjD.
Methods: Primary salivary gland epithelial cells (SGECs) were generated from minor salivary gland biopsies of SjD patients and controls. Expression of m 6 A machinery and mitochondrial RNA degradation pathway components was analyzed by public bulk and single-cell transcriptomics, quantitative PCR, immunoblotting, and immunofluorescence. Functional studies were performed using pharmacological inhibition or knockdown of the m 6 A writer METTL3. Endogenous dsRNA was detected by immunofluorescence. IFN responses and inflammatory mediators were assessed by RNA sequencing, qPCR, and ELISA.
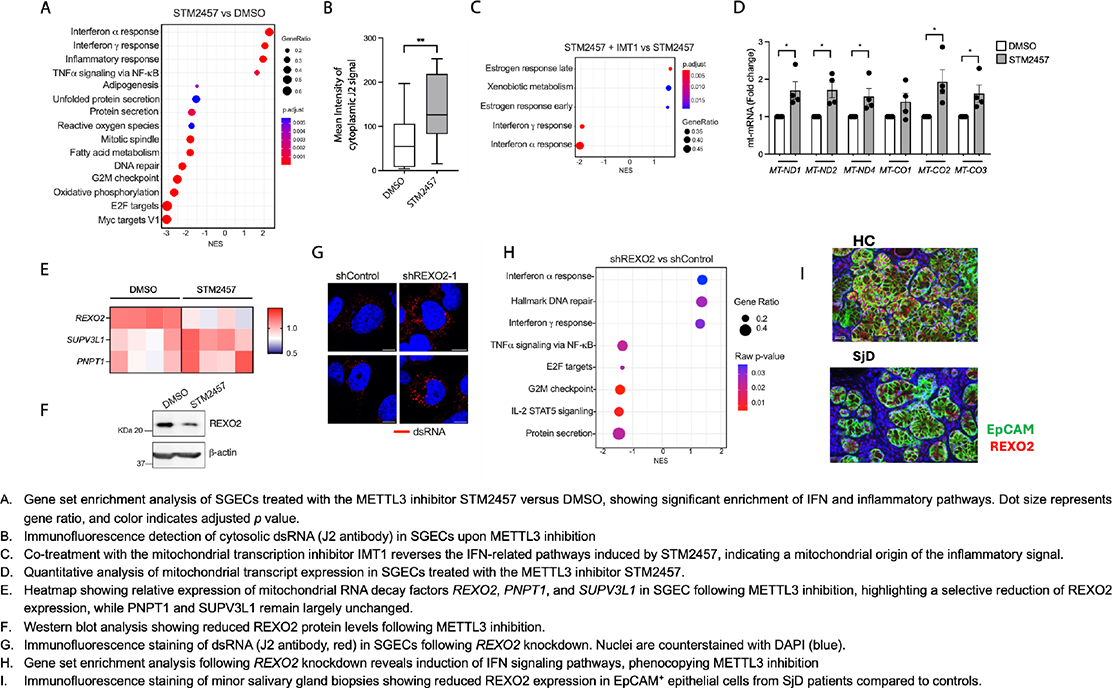
Results: Pharmacological inhibition of METTL3 in SGECs (using STM2457) induced a strong type I and type II IFN transcriptional program, as revealed by pathway enrichment analysis (Figure 1A). This response was associated with a marked intracellular accumulation of endogenous dsRNA (Figure 1B). Co-treatment with the mitochondrial transcription inhibitor IMT1 significantly reduced dsRNA accumulation and attenuated the IFN response, indicating a mitochondrial origin of the immunostimulatory RNA (Figure 1C, D). Analysis of mitochondrial RNA turnover pathways showed that while expression of the mitochondrial degradosome components PNPT1 and SUPV3L1 was unchanged, expression of the mitochondrial exonuclease REXO2 was significantly reduced following METTL3 inhibition (Figure 1E, F). Importantly, REXO2 knockdown in SGEC was sufficient to induce intracellular dsRNA accumulation and an IFN-driven transcriptional signature, closely mimicking the effects of METTL3 inhibition (Figure 1G, H). Consistently, immunofluorescence analysis of minor salivary gland biopsies revealed reduced REXO2 expression in EpCAM + epithelial cells from SjD patients compared with controls (Figure 1I).
Conclusions: These findings suggest that defective m 6 A-dependent epitranscriptomic regulation in epithelial cells leads to impaired mitochondrial RNA surveillance, resulting in accumulation of endogenous mitochondrial dsRNA and chronic IFN activation. This mechanism may explain the IFN signature observed in SjD and potentially other autoimmune diseases. Loss of REXO2 emerges as a key epithelial checkpoint in this pathway, and restoring mitochondrial RNA degradation may represent a novel therapeutic strategy to limit pathogenic interferon responses.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.