fetching data ... 
Background: T helper 17 (T H 17) cells have been heavily implicated in the pathogenesis of autoimmune and autoinflammatory disease. They play pivotal roles in driving proinflammatory responses by producing the key effector cytokine such as IL-17. T H 17 cell programming is controlled by epigenetic mechanisms including histone modifications [1,2]. In this context, clinical benefits from targeting T H 17 cells differentiation via epigenetic regulation have more promise rather than blocking single effector cytokines. However, supporting research was poorly conducted.
Objectives: Among rheumatic diseases, T H 17 cells are particularly important in Ankylosing Spondylitis(AS), and IL-17 inhibition is known to be good options for treatment in AS. Here we aimed to decipher the function of NSD1, Histone 3 lysine 36 (H3K36) mono- or di-methyl transferase in T H 17 cell differentiation and AS pathogenesis. Furthermore, we evaluated the potential of NSD1 inhibitor (BT5) as a clinical applicable candidate using human spinal tissues and AS animal model.
Methods: For in vitro study, isolated naïve CD4+ T cells from mouse spleen were cultured under specific conditions to induce differentiation into T H 17 cells. The cells were treated with a specific T H 17-inducing cytokine cocktail followed by treatment with BT5 or vehicle; infection of Lenti-Cas9 or -dCas9 with NSD1 specific sgRNA (sgNSD1). The differentiated T H 17 cells were analyzed by flow cytometry, RT-PCR, Western-blotting and ChIP-qPCR. BT5-treated osteoprogenitor cells derived from patients with AS were examined by RNA-seq and imaging analysis. For in vivo study, Curdlan-administered SKG mice were treated with BT5 (i.p., 10 mg/kg, 1 time/3 days) during 3 weeks. Inguinal and popliteal nodes were analyzed by flow cytometry. Blood samples were used to assess hepatotoxicity and nephrotoxicity. Bones of the ankle and foot were analyzed by micro-CT. Bioinformatic analysis allowed to see direct binding of NSD1 and changes in its target residue related to T H 17-specific gene loci.
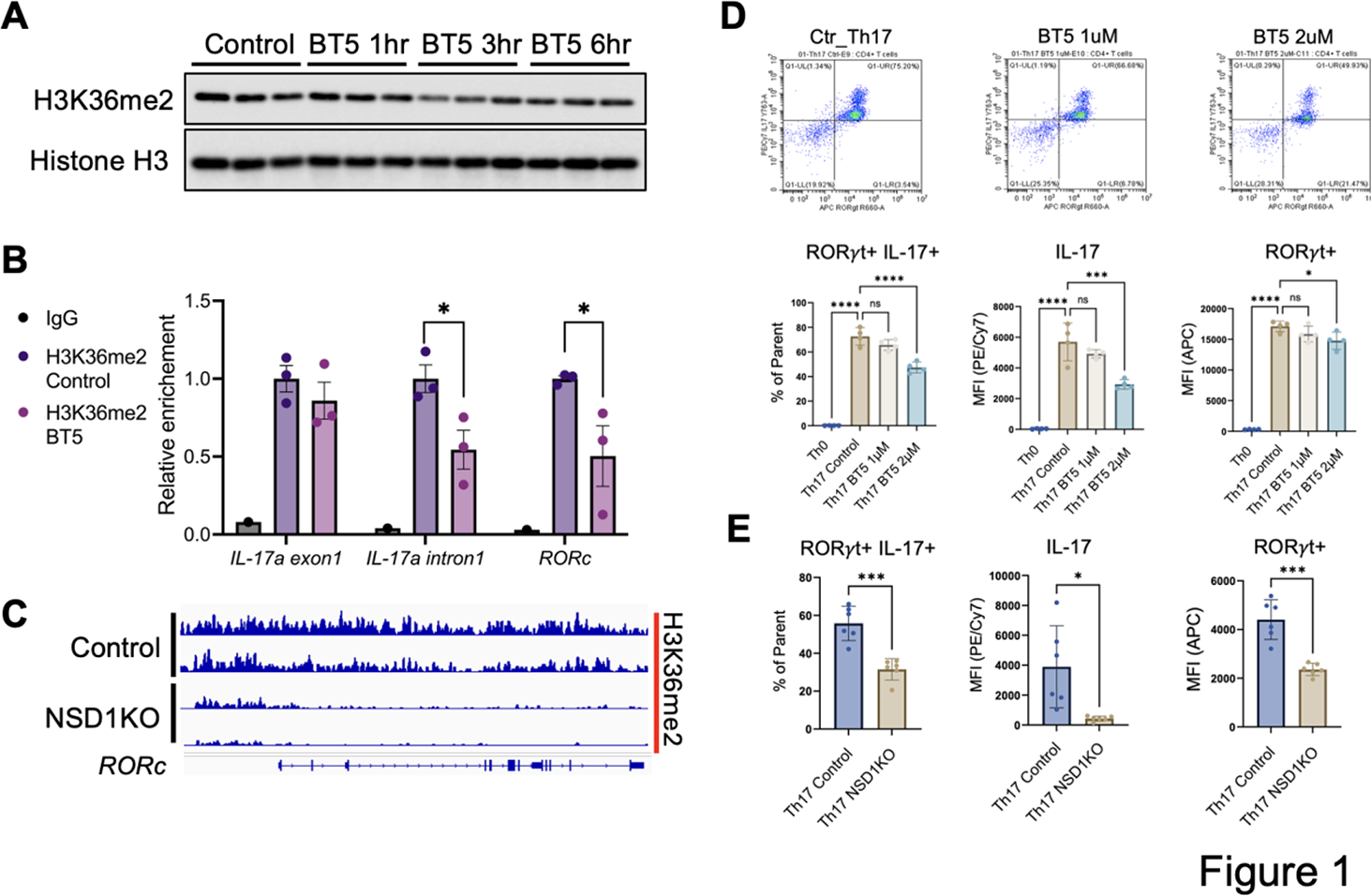
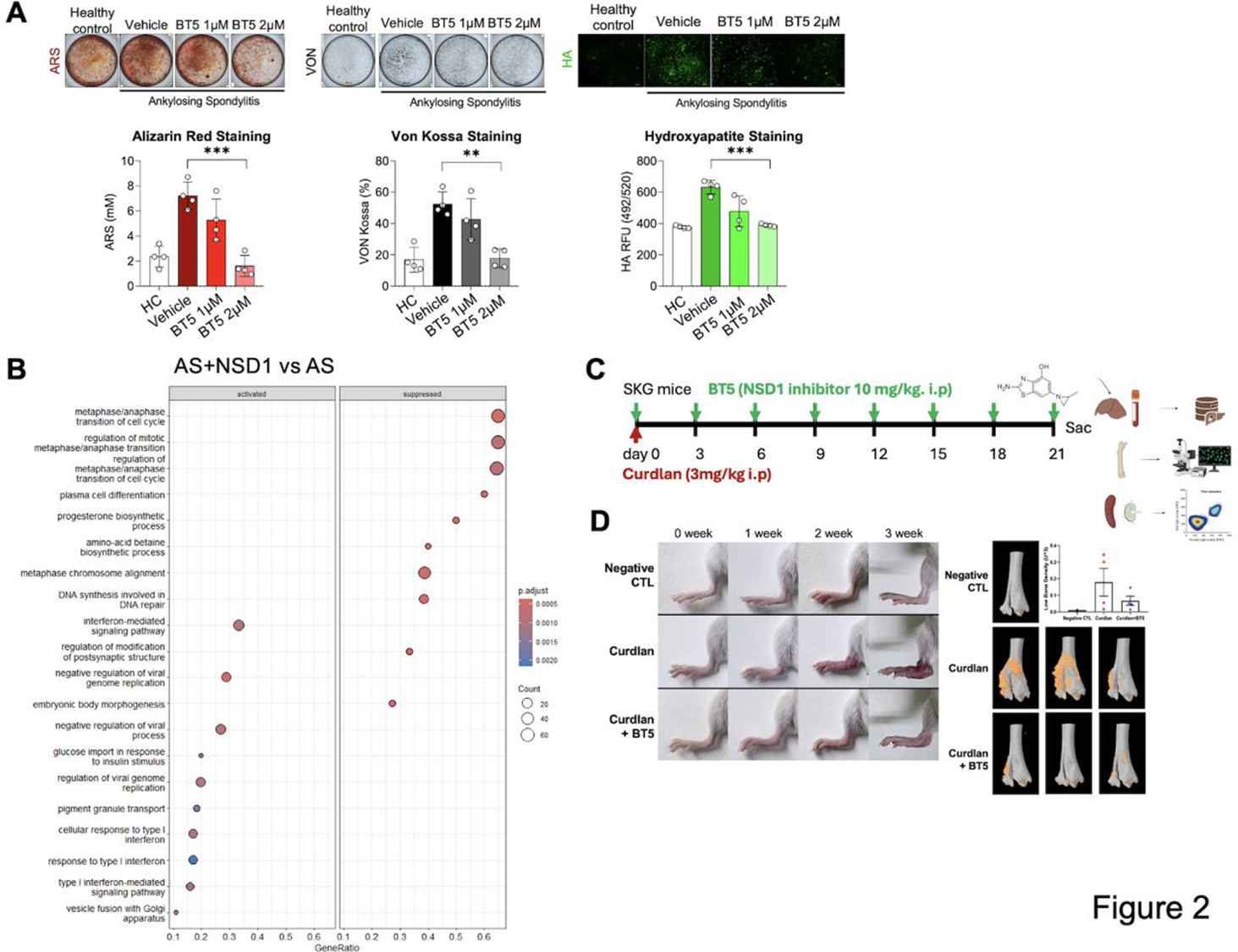
Results: BT5 treatment reduced H3K36me2 levels in mouse splenocytes and interrupted deposition of H3K36me2 in IL-17 and RORc (RORgt), after 3-hour treatment (Figure 1A, 1B). In line with this finding, inactivation of NSD1 by CRISPR/Cas9 editing resulted in reduction of H3K36me2 level at RORc locus in mouse mesenchymal stem cells (Figure 1C). BT5 or Cas9 with sgNSD1 inhibited T H 17 differentiation by attenuating expression of RORgt with decreasing IL-17 in mouse CD4+ T cells (Figure 1D, 1E). NSD1 inactivation also reduced matrix mineralization in AS-osteoprogenitor cells (Figure 2A), following IFN-mediated signaling pathway delaying osteoblast differentiation (Figure 2B) [3,4]. Following the indicated treatment scheme (Figure 2C), BT5 treatment attenuated inflammatory response in lymph nodes and ankles of SKG mice (Figure 2D).
Conclusions: In this study, we evaluated therapeutic efficacy of BT5 using in vivo model of chronic inflammation. Our molecular studies support that NSD1 inhibitor has a potent at reducing inflammatory response via directly dysregulation of specific gene loci, contributing to development of autoinflammatory disease. Our findings from AS-osteoprogenitor cells and mouse models provide promising evidence that NSD1 is a valid therapeutic target to suppress expressions of proinflammatory cytokines and T H 17 differentiation.
REFERENCES: [1] Jay A, Pondevida CM, Vahedi G. The epigenetic landscape of fate decisions in T cells. Nat Immunol. 2025 Apr;26(4):544-556. doi: 10.1038/s41590-025-02113-x. Epub 2025 Mar 19. PMID: 40108419.
[2] Park E, Ciofani M. Th17 cell pathogenicity in autoimmune disease. Exp Mol Med. 2025 Sep;57(9):1913-1927. doi: 10.1038/s12276-025-01535-9. Epub 2025 Sep 1. PMID: 40887501; PMCID: PMC12508148.
[3] Woeckel VJ, Eijken M, van de Peppel J, Chiba H, van der Eerden BC, van Leeuwen JP. IFNβ impairs extracellular matrix formation leading to inhibition of mineralization by effects in the early stage of human osteoblast differentiation. J Cell Physiol. 2012 Jun;227(6):2668-76. doi: 10.1002/jcp.23009. PMID: 21898404.
[4] Hayashida C, Ito J, Nakayachi M, Okayasu M, Ohyama Y, Hakeda Y, Sato T. Osteocytes produce interferon-β as a negative regulator of osteoclastogenesis. J Biol Chem. 2014 Apr 18;289(16):11545-11555. doi: 10.1074/jbc.M113.523811. Epub 2014 Mar 7. PMID: 24610813; PMCID: PMC4036289.
Acknowledgments: NIL.
Disclosure of Interests: None declared.