fetching data ... 
Background: Genome-wide association studies have shown that ~80% of rheumatoid arthritis (RA) risk variants lie in non-coding regulatory elements, which poses a challenge for determining their functional roles and cell type-specific contributions to disease susceptibility. While our epigenetic profiling previously showed that a substantial portion of these risk loci are active within synovial fibroblasts (SFs) [1], the specific gene programs they perturb and the contexts in which they operate remain largely undefined.
Objectives: We aimed to integrate RA risk variants with SF-specific regulatory regions and characterize their responsiveness to RA-relevant cytokines, thereby identifying distinct functional risk modules and mapping their activity to specific synovial fibroblast niches.
Methods: From the previously identified genetic risk regions, SNPs with posterior probability ≥0.01 were retained, and genes within 50 kb of each SNP or Capture Hi-C interaction anchors (unstimulated and TNF-stimulated from cultured RA SFs) were considered candidate risk genes. Actively expressed genes were identified using in-house [1] (n=20) and public [2] RNA-seq data (n=30) from cultured RA SFs. For eQTL analysis in unstimulated samples we used normalized RNA-seq levels regressed on SNP dosage with sex as covariate, meanwhile TNF-stimulated samples were tested with Kruskal-Wallis, with FDR ≤0.05 being significant.
Stimulus-specific responses were calculated as expression changes relative to unstimulated condition and Z-score–scaled per gene across stimulations using a public RNA-seq dataset [2]. Genes were clustered by hierarchical clustering (Euclidean distance, Ward’s method) into four clusters. Differential expression significance was obtained using DESeq2 (BH-adjusted p < 0.05). For each cluster and stimulus, mean Z-scores and the fraction of significant genes were calculated. Enrichment of SF-specific risk loci genes within clusters was tested using chi-square with Pearson residuals (|residual| > 2) indicating significance. Modules were refined using in-house synovial scRNA-seq [3]. (RA=13, non-inflamed controls=6) by retaining only genes with stimulus-consistent significant regulation in RA fibroblasts. These refined sets were scored across fibroblast clusters and diseases using AddModuleScore ( Seurat ).
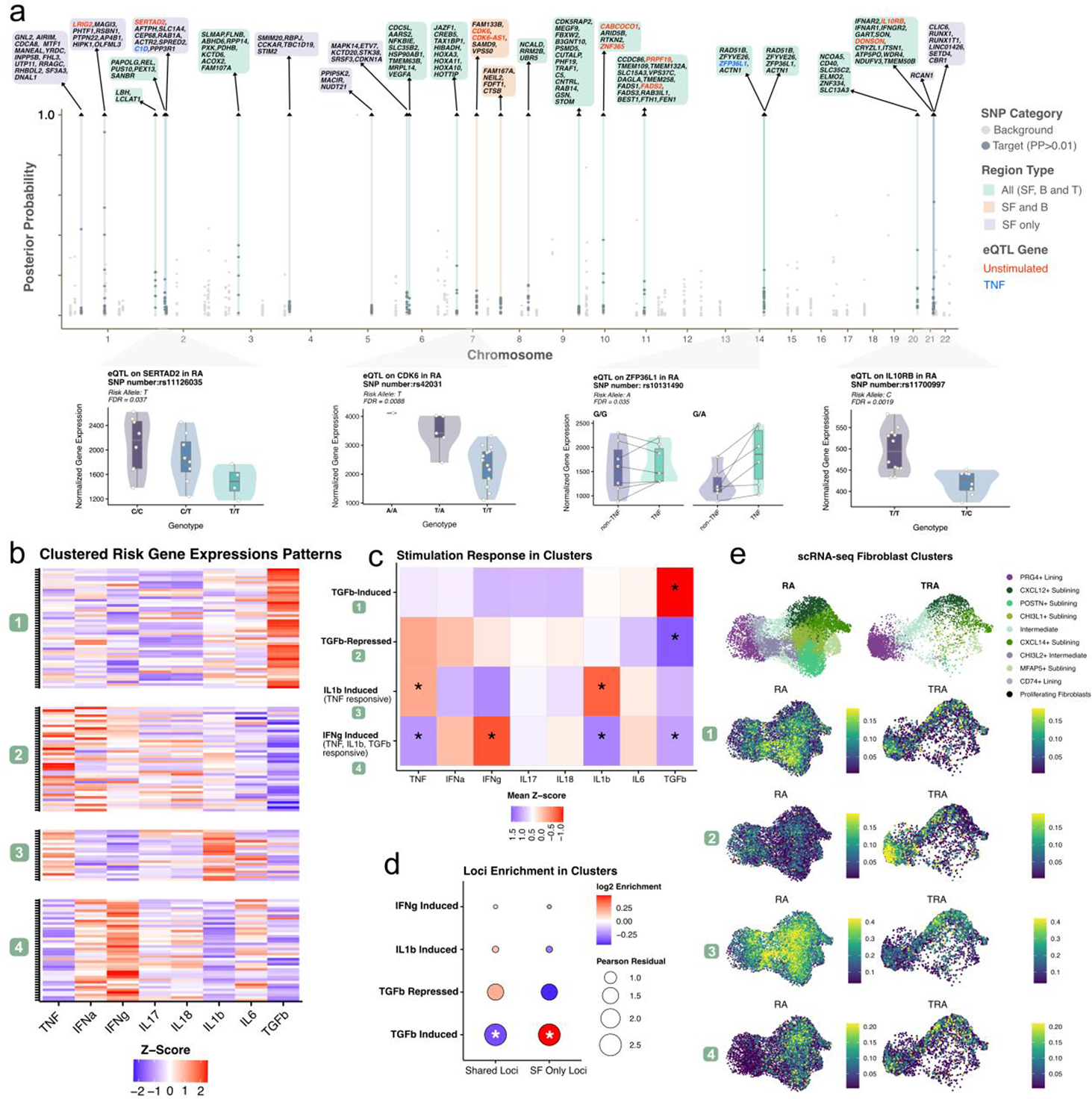
Results: Out of the 73 non-HLA RA genomic risk regions, 23 (31%) overlapped with active regulatory regions in SFs [1]. Of these, 8 regions were selectively open and active in SFs, whereas 15 were shared regulatory regions also accessible in B and T cells. By integrating chromatin interactions and transcriptional activity, we identified 150 candidate RA SF-risk genes (Figure 1a, top). eQTL analysis in unstimulated and TNF-stimulated SFs revealed ten genes across six risk loci with significant eQTLs in the unstimulated condition, including SERTAD2 , CDK6 , and IL10RB and two genes, ZFP36L1 and C1D , across 2 risk loci displayed TNF-dependent eQTLs (Figure 1a, bottom). This indicated, however, that additional stimuli are required for functional manifestation of certain genetic effects on gene expression in SFs. To systematically characterize the cytokine responsiveness of the 150 RA SF–risk genes, we performed hierarchical clustering based on transcriptional responses to RA-relevant cytokines. With this analysis we identified four gene modules (Figure 1b). Quantification using mean z-scores and the proportion of significantly regulated genes (>50% threshold) revealed that Module 1 was predominantly TGFβ-induced, Module 2 TGFβ-repressed, Module 3 IL-1β-induced with additional TNF responsiveness, and Module 4 IFNγ-induced with broader responsiveness to TNF, IL-1β, and TGFβ (Figure 1c). We next assessed whether genes residing in SF-specific regulatory regions were preferentially enriched in particular modules. Chi-square testing demonstrated significant enrichment of SF-specific loci genes in the TGFβ-induced module, while genes from shared B/T cell-SF loci were significantly depleted from this module (Figure 1d). Finally, we assessed the gene clusters using module scoring with in-house scRNA-seq data of synovial tissue, focusing on SF subpopulations. (Figure 1e). The TGFβ-induced module was more active in RA SFs compared to non-inflammatory controls, with strongest enrichment in RA-associated sublining populations including POSTN + and CHI3L1 + sublining fibroblasts. In contrast, the TGFβ-repressed module showed preferential activity in PRG4 + lining fibroblasts in control samples but it was highly reduced across fibroblast populations in RA.
Conclusions: By integrating RA GWAS loci with chromatin interactions and eQTL data in SFs, we identified 150 candidate SF risk genes and show that RA genetic risk in SFs may preferentially affect cytokine-responsive regulatory programs. In particular, SF-specific RA risk genes tend to drive a TGFβ-induced program in pathogenic RA fibroblast subpopulations, whereas homeostatic TGFβ-repressed programs in lining fibroblasts appear to be diminished in RA.
. (a, top ) Manhattan plot of non-HLA RA risk regions; SF-active genes are labeled (significant eQTLs colored). (a, bottom ) Representative eQTLs in unstimulated ( SERTAD2, CDK6, IL10RB ) and TNF-stimulated ( ZFP36L1 ) conditions. (b ) Hierarchical clustering of 150 RA SF-risk genes based on transcriptional responses to RA-relevant cytokines. (c ) Mean z-scores per module with * indicating >50% of genes significantly regulated. (d ) Enrichment of SF-specific versus shared B/T cell-SF loci across modules. (e ) Module scores projected onto fibroblast subpopulations in scRNA-seq of synovial tissue.
REFERENCES: [1] Ge X, Frank-Bertoncelj M, Klein K, et al. Genome Biol . 2021;22(1):247.
[2] Tsuchiya H, Ota M, Sumitomo S, et al. Ann Rheum Dis . 2021;80(4):440-450.
[3] Khmelevskaya A, Apostolopoulou K, Calvo Cebrian C, et al. bioRxiv. 2025. doi: 10.1101/2025.11.17.688477.
Acknowledgments: NIL.
Disclosure of Interests: None declared.