fetching data ... 
Background: Gout, a chronic hyperuricemia-induced disease causing joint damage and systemic complications, affects millions globally. Enhancing urate excretion remains a key yet challenging therapeutic strategy. Estradiol (E2) exhibits uricosuric effects, potentially mediated via estrogen receptors, including ERβ, though its precise role is unclear. The urate transporter GLUT9 is a critical reabsorption regulator and a promising therapeutic target. Prior evidence suggests E2 downregulates GLUT9 through ERβ via the PI3K/AKT1 pathway, but the underlying mechanism requires elucidation.
Objectives: This study aims to delineate how E2 promotes urate excretion via the ERβ/PI3K/AKT1/GLUT9 axis in gout.
Methods: This study aimed to investigate the mechanism by which estradiol (E2) promotes urate excretion. The expression of related genes in human kidney tissues was detected using immunohistochemistry (IHC), and RNA sequencing was employed to screen E2-targeted genes in the renal tubular epithelial cell line HK-2. Subsequently, a series of in vitro experiments were conducted to examine the role of the identified protein in the E2-ERβ-Glut9 pathway.Clinical Specimens. Fasting blood was collected from 26 controls and 24 hyperuricemic gout patients. Kidney tissues were from 3 controls and 3 patients. Clinical correlations were analyzed via Chi-square. Ethics approval (PYRC-2025-082-01) and informed consent were obtained.
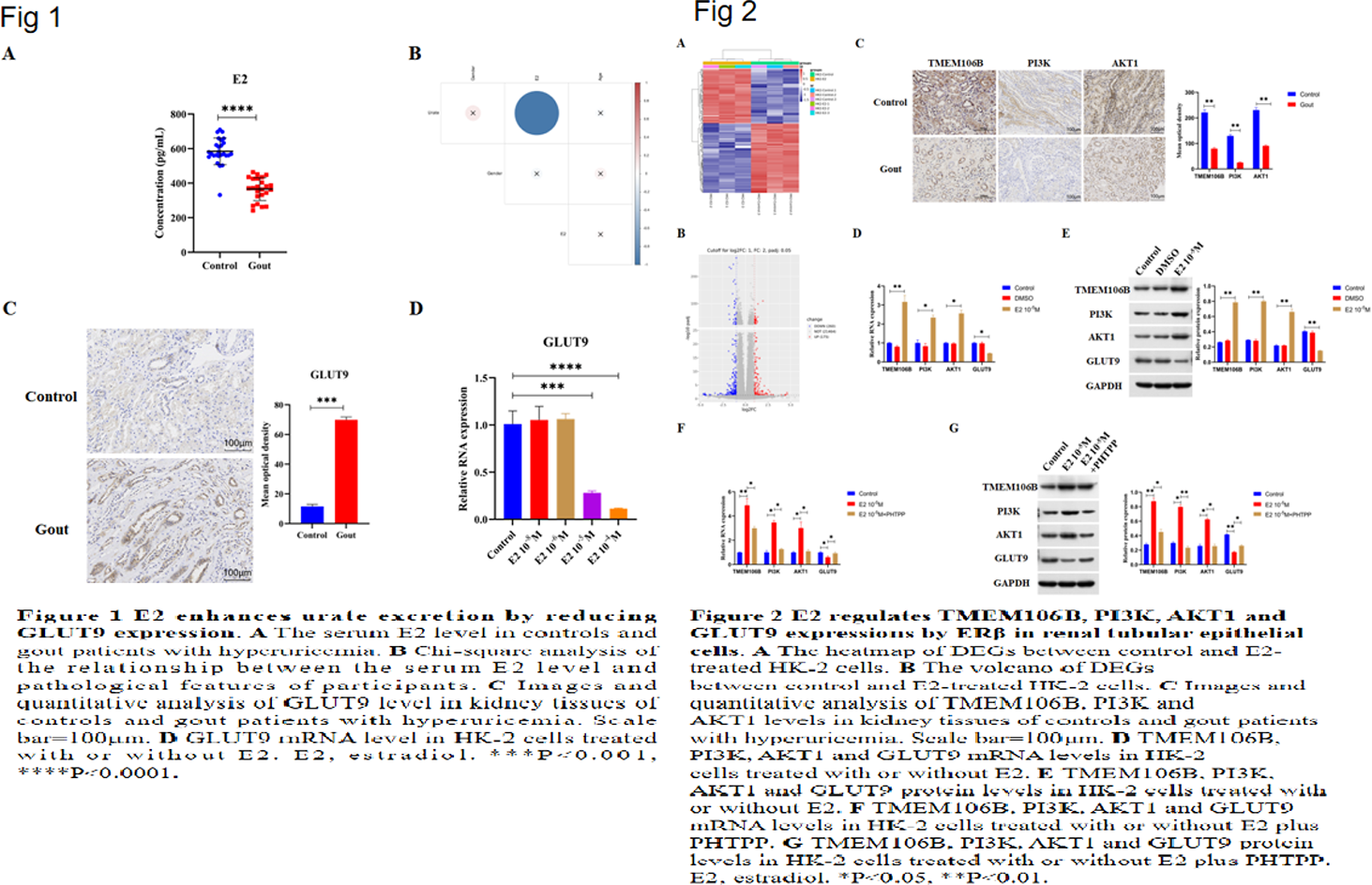
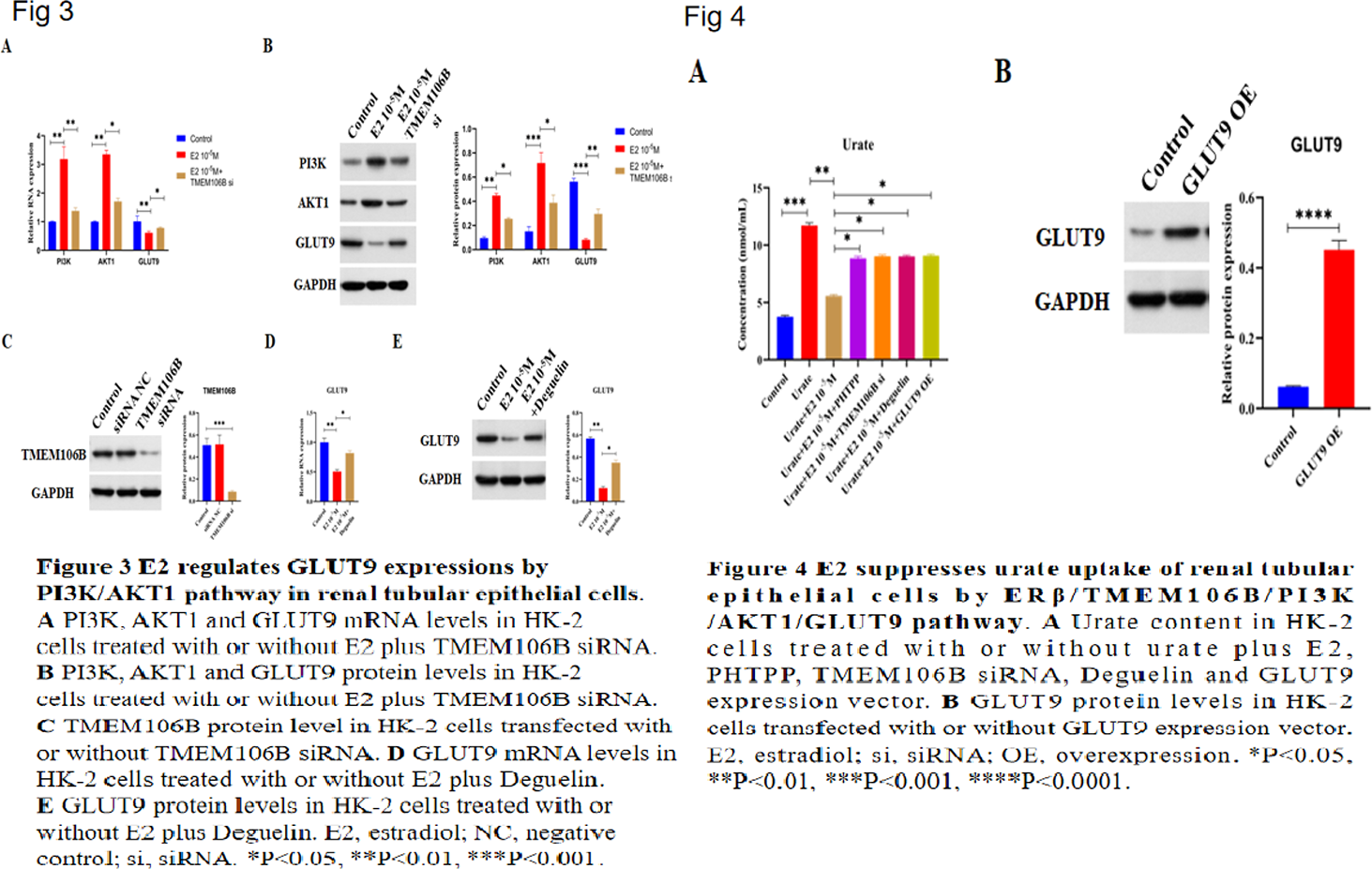
Results: E2 promotes urate excretion by downregulating GLUT9 Serum estradiol (E2) levels were measured in controls and hyperuricemic gout patients. E2 was significantly lower in gout patients and negatively correlated with serum urate (Figures 1A, 1B), suggesting estrogen promotes urate excretion. Immunohistochemistry showed elevated GLUT9 in patient kidneys (Figure 1C). In HK2 cells, E2 dosedependently reduced GLUT9 mRNA (Figure 1D), indicating E2 enhances urate excretion by downregulating GLUT9. E2 regulates TMEM106B, PI3K, AKT1 and GLUT9 via Erβ RNA sequencing of HK2 cells identified 175 up and 260 downregulated genes after E2 treatment (Figures 2A, 2B), including upregulated TMEM106B, a potential PI3K upstream gene. IHC revealed decreased TMEM106B, PI3K and AKT1 in gout kidneys (Figure 2C). E2 increased TMEM106B, PI3K and AKT1 but decreased GLUT9 in HK2 cells (Figures 2D, 2E). PHTPP (ERβ antagonist) reversed these effects (Figures 2F, 2G), indicating ERβdependent regulation. TMEM106B mediates E2 effects on PI3K/AKT1/GLUT9 E2 elevated PI3K and AKT1 while lowering GLUT9 in HK2 cells (Figures 3A, 3B). TMEM106B siRNA abolished these changes (Figures 3A–C), demonstrating that E2 modulates PI3K, AKT1 and GLUT9 through TMEM106B. PI3K/AKT1 pathway controls GLUT9 expression E2 reduced GLUT9 in HK2 cells (Figures 3D, 3E). Deguelin (PI3K/AKT1 inhibitor) blocked this reduction, confirming that E2 regulates GLUT9 via the PI3K/AKT1 pathway. E2 inhibits urate uptake via ERβ/TMEM106B/PI3K/AKT1/GLUT9 Urate treatment increased intracellular urate in HK2 cells, confirming active uptake. E2 suppressed this uptake (Figure 4A). Inhibition of ERβ (PHTPP), TMEM106B knockdown, PI3K/AKT1 inactivation (deguelin) or GLUT9 overexpression (Figure 4B) all reversed E2mediated suppression (Figure 4A). Thus, E2 reduces renal urate uptake through the ERβ/TMEM106B/PI3K/AKT1/GLUT9 axis.
Conclusions: These findings reveal that E2 promotes urate excretion by reducing GLUT9 expression via activating ERβ/TMEM106B/PI3K/AKT1 pathway in renal tubular epithelial cells, which provide novel targets and insights for gout treatment.
REFERENCES: [1] Baggen J, Persoons L, Vanstreels E et al (2021) Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat Genet 53, 435-444.
Acknowledgments: NIL.
Disclosure of Interests: None declared.