fetching data ... 
Background: Schnitzler’s syndrome (SchS) is a rare systemic auto-inflammatory disease combining monoclonal gammopathy, urticarial rash, inflammatory manifestations and bone involvement [1]. The dramatic efficacy of IL-1 blockade [2], combined with experimental data showing excessive IL-1β production by PBMCs [3], supports the key role of IL-1β in the pathophysiology of the disease. However, the causal link between monoclonal gammopathy and the inflammatory syndrome associated with IL-1 hyperproduction remains unexplained, as does the role of B-cell clones in the pathogenesis of Schörl’s syndrome.
Objectives: Considering the systematic presence of monoclonal gammopathy in SchS, the aim of the study was to identify genetic alterations and/or specific VDJ gene rearrangement in the CD19+ fraction of bone marrow from patients with SchS. We also sought to examine the impact of these mutations on B cells and whole bone marrow, particularly myeloid cells.
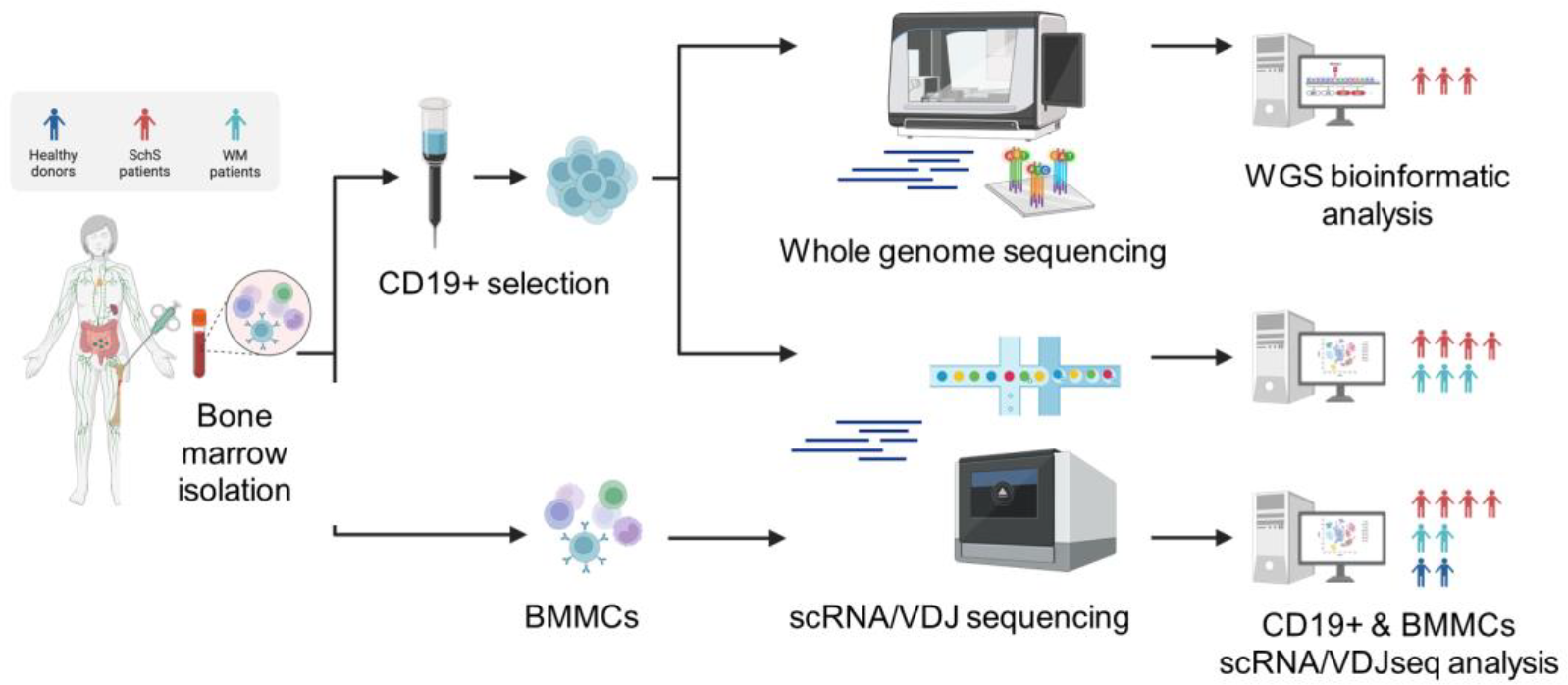
Methods: To address these questions, we conducted a multi-omic analysis of whole bone marrow and sorted CD19+ cells from 5 SchS patients, integrating whole genome sequencing, single-cell RNA and VDJ sequencing ( Figure 1 ). All 5 patients validated Strasbourg criteria (1) and had an IgM gammopathy. Bone marrow samples from healthy donors and patients with Waldenström macroglobulinemia (WM) were included as controls to obtain transcriptomic profiles of immune cell populations in a normal context and in another disease with a proliferation of B cell clones.
Results: Whole bone marrow single-cell data revealed a significant enrichment of T cells in SchS patients compared to healthy controls, but not compared to patients with WM. Analysis of more than 60,000 B cells showed no difference in total B cell count in SchS versus controls, but a marked shift toward more differentiated B cell stages (mature B and plasma cells), suggesting clonal expansion of mature B cells in SchS. Assuming an immunoglobulin pathogenesis, we searched for recurrent V(D)J gene segments. Some recurrences have been observed, but no single pathogenic immunoglobulin profile has been identified. However, while single-cell VDJ-seq revealed clonal B cell expansions in all SchS and WM patients, only SchS patients exhibited oligoclonality—with multiple coexisting B cell clones (median of 3 per patient, minimum clonal size of 2%)—in contrast to the monoclonality observed in WM. The clones in SchS were transcriptionally distinct from non-clonal B cells and showed heterogeneous differentiation states, suggesting specific gene expression programs. We therefore hypothesized that a recurrent mutation involving inflammatory pathways could explain both oligoclonality and autoinflammation. Using WGS to identify mutations, followed by scRNAseq to assess mutation expression, we were able to identify 3 recurrent mutations: MYD88, CXCR4, and LVRN. Myeloid differentiation primary-response gene 88 (MYD88) is a key adaptor molecule for the transduction of signals from IL-1 receptors and TLRs. MYD88 mutations were identified in all five patients: the hotspot L265P in SCH2–5 and also in SCH1 by scRNA-seq, plus an additional M219T mutation in SCH4. Truncating CXCR4 mutations occurred in 4/5 patients and were confined to specific clones: S338* in SCH4–5 (and also in SCH3 and SCH2 by scRNA-seq) plus a minor subclone S341Ffs*3 in SCH3. Two missense mutations of LVRN (Laverin, an aminopeptidase notably involved in human placentation) were encountered in two samples but not expressed in our scRNA-seq data arguing against a pathogenic role. Variant allele frequencies (VAFs) associated with single-cell read mapping allowed us to determine the proportion of B cells carrying each mutation and their association with specific B cell clones. All dominant clones harbored activating MYD88 mutations, whereas CXCR4 mutations are restricted to specific clones in SCH3 and SCH4. In addition, we did not detect large-scale chromosome copy-number alteration in SchS patients, nor recurrent structural rearrangements. Regarding differential gene expression, clonal B cells exhibited an overexpression of angiogenesis and inflammation pathways including IL6/JAK/STAT and NF-κB signaling, alongside a down-regulation of anti-inflammatory genes. B-cell progenitors showed enrichment in genes derived from inflammatory pathways (TNF pathway via NFKB), with a significant increase in TNF expression, particularly in ProB cells, compared to controls and Waldenström. Concomitantly, T cells of SchS patients displayed an upregulation of the NF-κB, interferon and RAGE signaling pathways compared to healthy controls, with an overproduction of pro-inflammatory cytokines such as TNF. Myeloid cells also upregulated inflammatory genes included alarmins, pattern recognition receptors, key NF-κB signaling components, and produced high levels of pro-inflammatory chemokines and vasoactive mediators. IL1B was not overexpressed in monocytes/granulocytes compared to control, indicating post-transcriptional mechanisms for high IL1B levels in patient serum. These changes reflect an increased inflammatory state of both T cells and myeloid cells from SchS patients, which may contribute to the systemic manifestations of the disease.
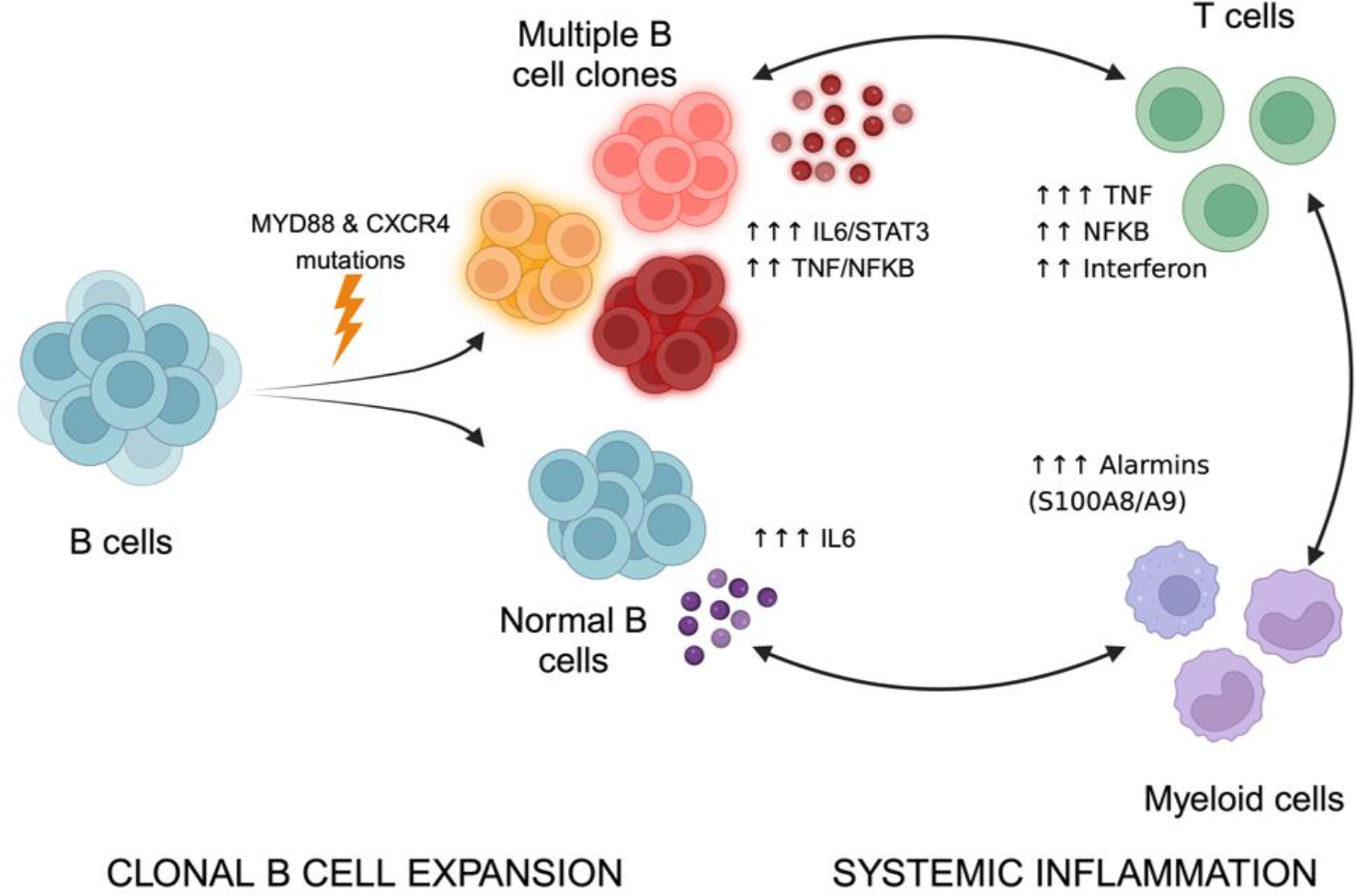
Conclusions: Overall, these data suggest a mechanism whereby early MYD88 and CXCR4 mutations appear to drive the expansion of multiple B cell clones that trigger a systemic inflammatory cascade, subsequently amplified by activated T cells and myeloid cells ( Figure 2 ).
Study workflow and analytical pipeline.
Proposed model linking MYD88/CXCR4-mutant clonal B-cell expansion to systemic inflammation
REFERENCES: [1] Simon A et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy 2013.
[2] Néel A et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome: a French multicenter study. Autoimmun Rev 2014.
[3] Pizzirani C et al. Dysfunctional inflammasome in Schnitzler’s syndrome. Rheumatology (Oxford) 2009.
Acknowledgments: NIL.
Disclosure of Interests: Stanislas Riescher Research grant 2024 Fond de dotation CSL Behring for the funding of multiomics analyses SOBI 3.000€ for multi-omics funding, Yannick Le Bris: None declared, Elise Douillard: None declared, Magali Devic: None declared, Mia Cherkaoui: None declared, Sébastien Barbarot: None declared, Florence Magrangeas: None declared, Stephane Minvielle: None declared, Eric Letouze: None declared, Antoine Néel Fond de dotation CSL Berhing, 20.000€ for multi-omics funding
SOBI 3.000€ for multi-omics funding.