fetching data ... 
Background: Still’s disease (SD) is a polygenic systemic autoinflammatory disorder with highly heterogeneous clinical features and disease course among patients. Therefore, genetic analyses are essential for elucidating pathogenic mechanisms and understanding clinical heterogeneity.
Objectives: This study aimed to explore potential variants associated with monogenic autoinflammatory diseases and to identify the characteristics of patients carrying variants, thereby contributing to a better understanding of the complex genetic mechanisms underlying SD.
Methods: Patients fulfilling the Yamaguchi criteria for SD and having available genetic data were included. Demographic, clinical, laboratory findings, and treatments were recorded using standardized forms. Anemia was defined as hemoglobin <12 mg/dL; neutrophilic leukocytosis as leukocyte count >10,000/µL (>80% neutrophils); elevated liver enzymes as values above the upper limit of normal; and hyperferritinemia as ferritin >3000 mg/dL. Macrophage activation syndrome (MAS) was evaluated according to the HLH-2004 criteria. Complete response was defined as the complete resolution of clinical signs and symptoms together with normalization of all relevant laboratory parameters. Partial response was defined as the persistence of clinical manifestations and/or laboratory abnormalities without full normalization. DNA isolated from peripheral blood was analyzed using the AmpliSeq® Library Kit Plus Autoinflammatory Disease Panel, targeting exons and exon–intron boundaries of 41 genes associated with autoinflammatory diseases. Target regions were amplified by PCR and sequenced using the Ion GeneStudio™ S5 Plus System (Ion Torrent™). Data were analyzed with Ion Reporter™ v5.20, and variants were interpreted using multiple databases (ClinVar, OMIM, dbSNP, gnomAD, ExAC) and classified according to the 2015 ACMG criteria.
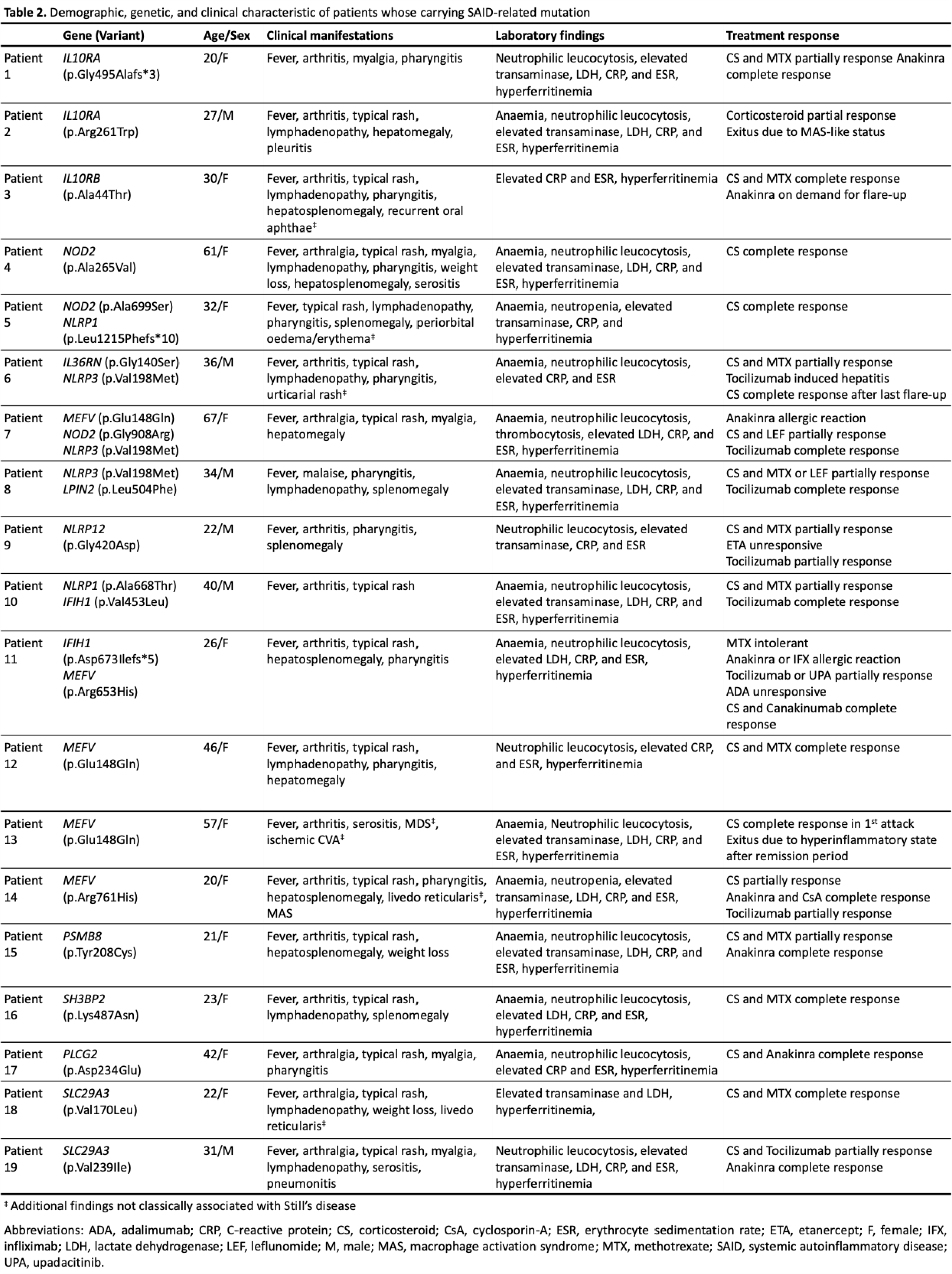
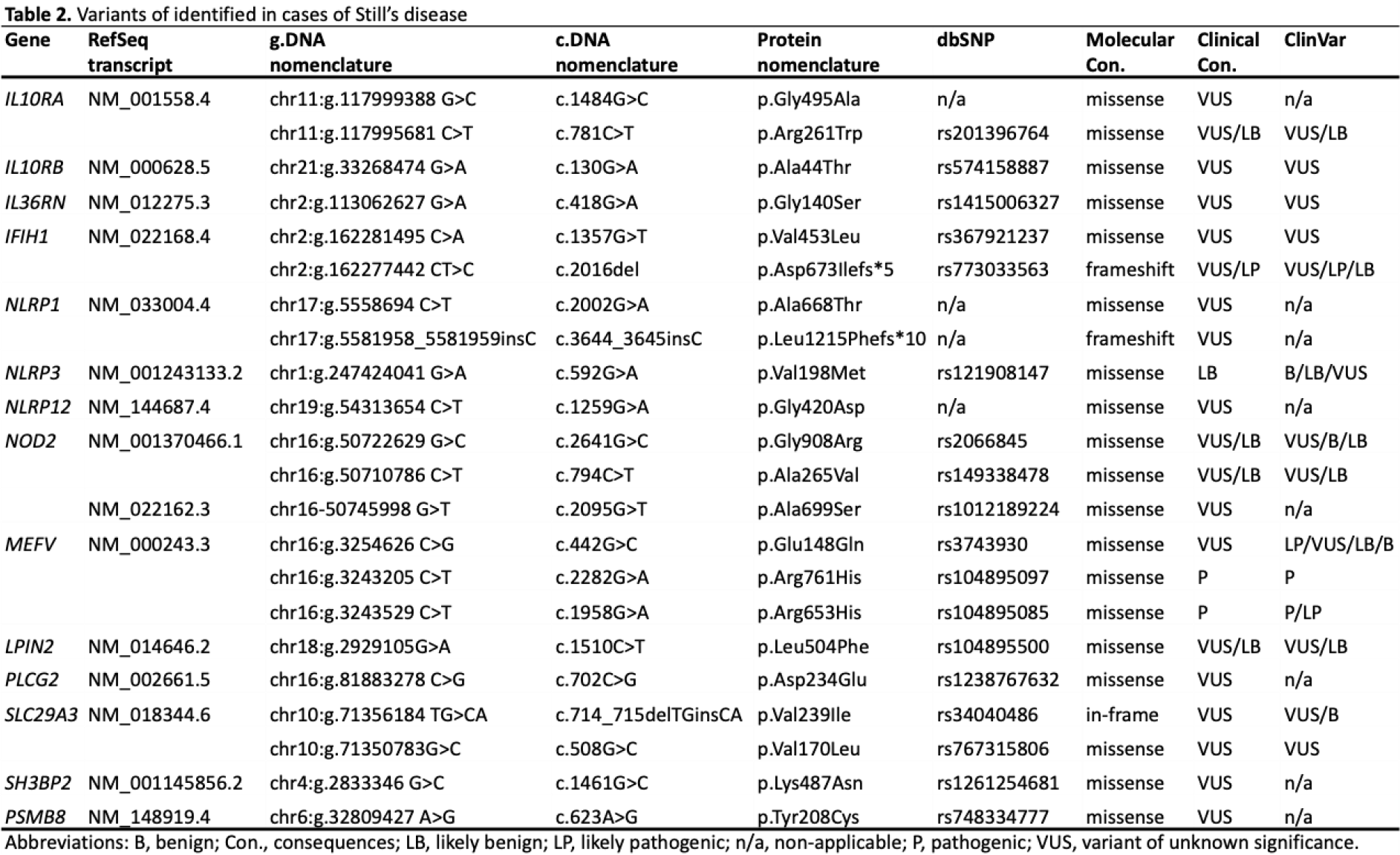
Results: A total of 108 patients were initially assessed for eligibility. Of these, 64 were excluded due to a lack of genetic testing, leaving 44 patients with available genetic data. Among these, 5 patients were excluded because of an undifferentiated diagnosis, 4 were reclassified as having another systemic autoinflammatory disease (SAID), and 1 was excluded due to missing data. The remaining 34 patients constituted the study cohort. The majority of patients were female (64.7%). The median age at diagnosis was 29.5 years (IQR: 22–40.5). Fever was present in all patients (100%). Arthralgia (79.4%) and rash (70.6%) were the most common clinical manifestations, followed by arthritis (55.9%), pharyngitis (52.9%), and lymphadenopathy (52.9%). Hepatomegaly and splenomegaly were observed in 35.3% and 32.4% of patients, respectively. Myalgia occurred in 35.3% of cases, while pleuritis and pericarditis were less frequent (17.6% and 14.7%, respectively). Weight loss was reported in 17.6% of patients. Laboratory findings demonstrated a high inflammatory burden. The median haemoglobin level was 11 g/dL (IQR: 9.72–12.2), and anaemia was present in 73.5% of patients. Median white blood cell and neutrophil counts were 14,500/mL (IQR: 11,948–19,963) and 11,930/mL (IQR: 9,177–16,897), respectively, with neutrophilic leucocytosis observed in 79.4% of cases. Abnormal liver transaminases were detected in 73.5% of patients, with median ALT and AST levels of 90 IU/L (IQR: 40–251) and 61 IU/L (IQR: 35–258), respectively. Median ferritin levels were markedly elevated at 9,067 ng/mL (IQR: 2,589–32,524), and hyperferritinemia was present in 76.5% of patients. Median CRP and ESR values were 199 mg/L (IQR: 118–260) and 75 mm/h (IQR: 60–86), respectively. Regarding treatment, almost all patients received corticosteroids (97.1%). Methotrexate was used in 58.8% of patients, while biologic therapies were commonly prescribed, including anti-IL-1 agents (55.9%) and anti-IL-6 agents (38.2%). Other treatments included leflunomide (11.8%), cyclosporine A (8.8%), anti-TNF agents (8.8%), and Janus kinase inhibitors (8.8%). In subgroup analyses, pericarditis was absent among patients without genetic variants but was observed in 5 of 19 variant carriers (26.3%). Similarly, pleuritis occurred less frequently in the non-carrier group (6.7%) compared with patients carrying variants (26.3%). Hyperferritinemia was more frequent in patients with the genetic variant (89.5%) than in non-carriers (60%); however, these differences did not reach statistical significance. Among the 19 patients carrying SAID–related variants, the detected variants were distributed across multiple genes, including IL10RA , IL10RB , IL36RN , IFIH1 , NLRP1 , NLRP3 , NLRP12 , NOD2 , MEFV , LPIN2 , PLCG2 , SLC29A3 , SH3BP2 , and PSMB8 , reflecting substantial genetic heterogeneity. MEFV , NLRP3 , NOD2 , and IFIH1 variants appeared most frequently, and several patients carried more than one variant concurrently. All mutations were heterozygous except for one patient with a homozygous SLC29A3 variant. The characteristics of patients carrying SAID–related genetic variants are presented in Table 1 , and detailed features of variants are presented in Table 2 .
Conclusions: A significant proportion of patients with SD carried variants in genes associated with autoinflammatory diseases. The predominance of variants of uncertain significance highlights the challenges in interpreting genetic findings. Long-term follow-up is needed to determine the clinical implications of these variants in SD. Genetic investigation may provide valuable insights into both diagnosis and differential diagnosis of SD, as well as other systemic autoinflammatory disorders. In addition to their effect on the inflammatory burden in the pathogenesis of disease, these variants may be epiphenomena. Larger patient cohorts and functional studies are required to clarify the clinical relevance of the detected variants.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.