fetching data ... 
Background: Immune checkpoint inhibitors (ICIs) are monoclonal antibodies that enhance anti-tumour immunity. They have revolutionised cancer outcomes, but immune-related adverse events (irAEs) that clinically resemble “spontaneous” immune-mediated inflammatory diseases (IMIDs) occur unpredictably, often leading to hospitalisation. They may be sustained or otherwise life-altering. Among them, ICI-induced inflammatory arthritis (ICI-IA) develops in ≥6% of recipients. Activation and trafficking of cytotoxic T cells to tissue may be a feature of irAE development [1,2], but risk factors for the phenomenon and mechanisms of tissue pathobiology remain poorly understood.
Objectives: Through prospective peripheral blood (PB) immune monitoring of an unselected, single centre inception cohort of ICI recipients, together with single-cell genomic profiling of paired PB and synovial tissue (ST) from incident rheumatoid arthritis (RA) patients, we sought predictive biomarkers of toxicity and related pathophysiological insight.
Methods: ICI recipients were recruited into the Monitoring immunE Dysregulation following Immune checkpOint inhibitioN (MEDALLION) inception cohort [3], undergoing serial peripheral blood sampling during ICI treatment for 10-months or until they developed a significant-irAE (sirAE; defined as grade > 3 CTC irAE affecting any tissue, clinically significant dysthyroidism, vitiligo or inflammatory arthritis). Two optimised spectral flow cytometry panels (28-marker and 10-marker) were applied, and data acquired using a Cytek Aurora. Unsupervised clustering enabled granular annotation of circulating T-cell populations, with observations confirmed by manual gating. Proteomic profiling of serially obtained paired sera was performed using the NULISAseq™ Inflammation Panel 250. In parallel, PB and paired ST were obtained from patients with ICI-IA and disease controls with RA – all of whom were naïve to glucocorticoids or disease-modifying antirheumatic drugs. After enzymatic disaggregation [4] and live cell sorting, 5’ single-cell RNA-Sequencing (scSeq) was undertaken using the 10X platform, applying gene signature scoring and interaction strength analyses following standard processing (UCell, CellChat). Paraffin-embedded tissues also underwent immunofluorescence staining for 60 cellular markers before imaging using the MACSima multiplex platform. All human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents under a REC approved protocol.
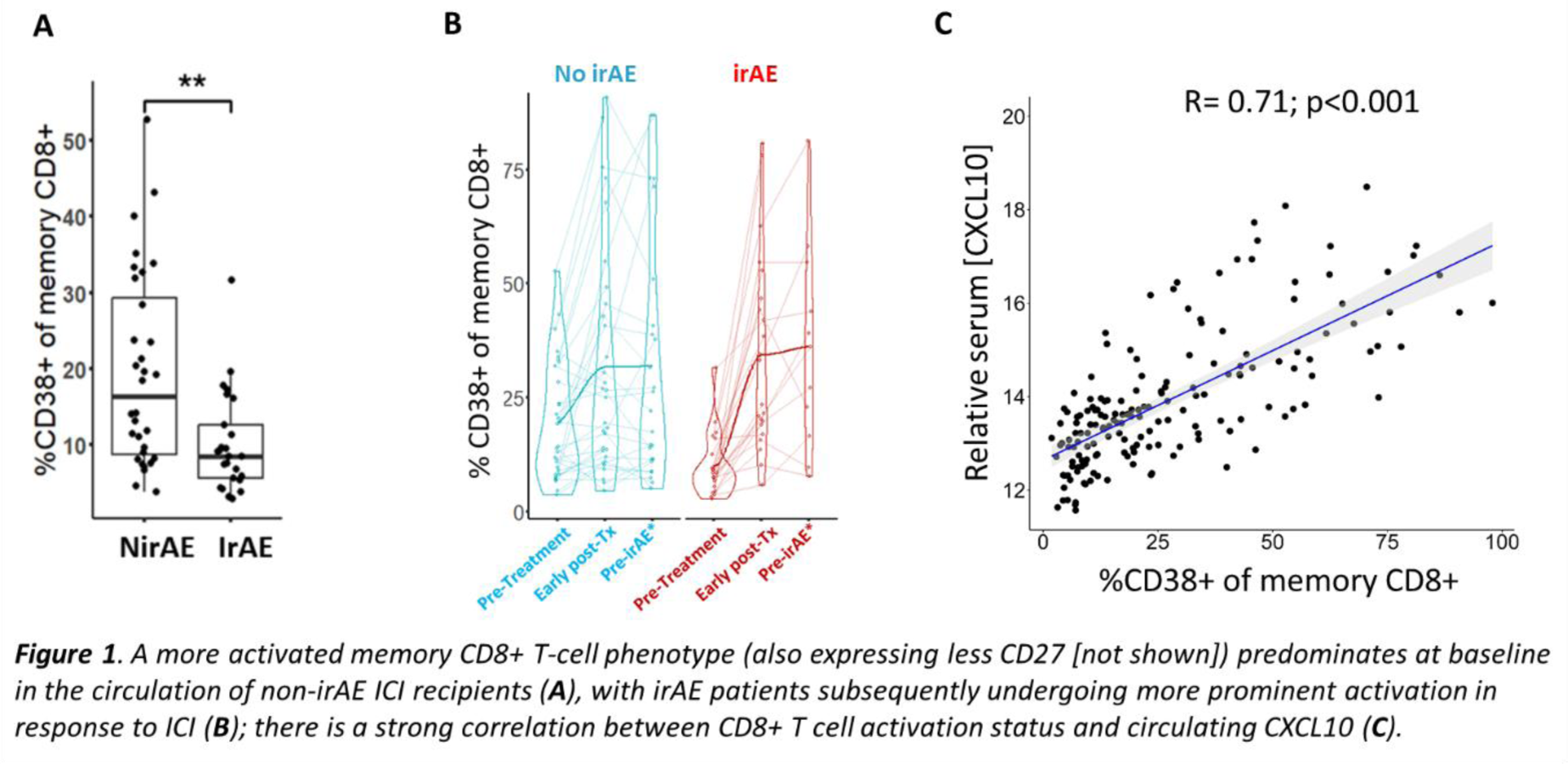
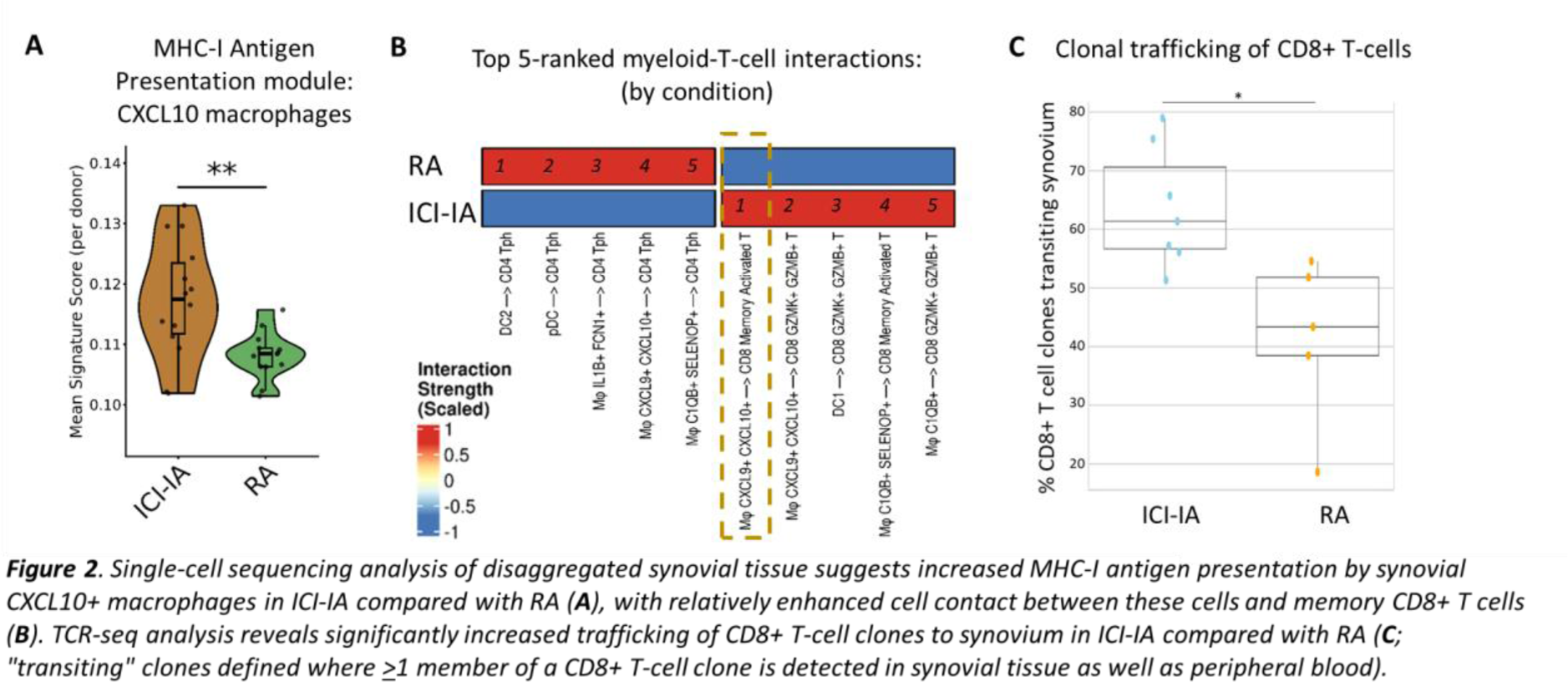
Results: 25 (44%) MEDALLION patients experienced ≥1 sirAE during the 10-month follow-up period, the only baseline clinical predictor of this outcome being treatment that included anti-CTLA-4 in combination with anti-PD-1 or anti-PD-L1 therapy (p=0.01; Chi-squared test). As expected, a higher proportion of participants experiencing sirAEs had clinical cancer responses to ICI therapy by close of follow-up compared with non-sirAE patients (72% versus 50%). Flow cytometry analysis showed a significantly heightened pre-treatment state of activation/exhaustion amongst circulating memory CD8+ T cells of ICI recipients who did not develop irAEs compared with those who did, with significantly increased CD38 expression ( Figure 1A ) and reduced CD27 expression; these characteristics were associated with unfavourable cancer outcomes. Subsequent ICI exposure led to significantly greater induced activation of circulating memory CD8+ T cells in irAE patients as determined by CD38 expression ( Figure 1B ). Of 250 serum protein analytes, the chemokine CXCL10 displayed the greatest relative increase over time from baseline, following treatment initiation, in sirAE patients. Intriguingly, given known up-regulation of the CXCL10 receptor CXCR3 on activated CD8+ T cells 2 , a striking correlation was also observed between circulating CXCL10 levels and CD8+ T-cell activation in the MEDALLION cohort ( Figure 1C ). We therefore hypothesised that migration of activated memory CD8+ T cells along a CXCL10 gradient to interact with synovial macrophages might represent a potential mechanism distinguishing ICI-induced from “spontaneous” IMIDs. Consistent with this, scSeq analysis of 197,654 ST cells from 15 ICI-IA and 13 RA patients demonstrated relative enrichment of an MHC-I antigen presentation gene signature amongst CXCL10+ macrophages of ICI-IA ST compared with RA patients ( Figure 2A ). Moreover, inferred cell contacts between CXCL10+ macrophages and activated memory CD8+ T cells were the most dominant myeloid-T-cell interactions in ICI-IA ST, in stark contrast to the situation in RA ST ( Figure 2B ). MACSima spatial proteomic analysis confirmed prominent interaction of these cell types in ICI-IA synovial sections (not shown). Finally, T-cell receptor (TCR) repertoire analysis of paired PB and ST T cells evidenced significantly increased trafficking of CD8+ T-cell clones to synovium in ICI-IA compared with RA ( Figure 2C ), with more granular analysis ongoing.
Conclusions: We propose systemic pre-treatment of cytotoxic T cell exhaustion predicts blunted ICI-induced activation; whilst this apparently shields non-malignant tissue from chemokine-mediated infiltration by activated pathogenic memory CD8+ T cells to support unwanted inflammation, it may also account for inferior cancer response outcomes. Our work points to more personalised approaches to immunotherapy based on predicted efficacy, and should inform therapeutic strategies in ICI-IA.
REFERENCES: [1] Wang R 2023 Science Immunol.
[2] Kim et al 2022 Nat Commun.
[3] Gault et al 2024 BMC Cancer.
[4] Donlin et al 2018 Arthritis Res Ther.
Authors AKM, AT, JDT and KW contributed equally.
Authors AGP and AF contributed equally.
Acknowledgments: NIL.
Disclosure of Interests: Anthony K. McLean: None declared, Amanda Thomson: None declared, Jason D. Turner: None declared, Kristian Williams: None declared, Abigail C Gault: None declared, Abbie Degnan: None declared, Joe Berry: None declared, Linda Hogarth: None declared, Julie Diboll: None declared, Ioana Nicorescu: None declared, Luke J Jones: None declared, George Merces: None declared, Ana Teodosio: None declared, Bethany Hunter: None declared, Imogen Wilson: None declared, Alison Bridgewood: None declared, Lisa-Jayne Brown-Schofield: None declared, Jeremie Nsengimana: None declared, Shashank Jariwala GSK, GSK, Qin An GSK, GSK, Lalit Pallan: None declared, Benjamin A. Fisher Novartis, Servier. Otsuka, Novartis, BMS, Servier, Galapagos, Roche, UCB, Sanofi, Johnson & Johnson, AstraZeneca, Otsuka, Amgen, Kiniksa, Cullinan, Quell, OneFour Bio, Johnson & Johnson, Servier, Galapagos, Novartis, Cullinan, Jonathan Coxhead: None declared, Rafiqul Hussain: None declared, Michal Magid-Slav GSK, David McDonald: None declared, Stefan Siebert Amgen, Johnson & Johnson, Novartis, Pfizer and UCB, AbbVie, AstraZeneca, Johnson & Johnson, Syncona, Shattuck Labs, Teijin Pharma, and UCB, Eli Lilly, GSK, Johnson & Johnson, Pfizer and UCB., Andrew Filby: None declared, Adam P. Croft: None declared, John Isaacs: None declared, Gary Reynolds: None declared, Ruth Plummer: None declared, Catharien Hilkens: None declared, David F. Tough GSK, Amy E Anderson: None declared, Andrew Filer Johnson & Johnson, Sonoma, Quell Therapeutics, Synact, GSK, Sonoma, Johnson & Johnson, Arthur Pratt GSK.