fetching data ... 
Background: Osteoarthritis (OA) is a chronic, low-grade inflammatory disease in which an increased M1/M2 macrophage ratio in the synovium accelerates cartilage degeneration and exacerbates disease progression. Genome-wide association studies implicate ASAP2 in OA risk, whereas its mechanistic role remains unclear.
Objectives: To identify the role of ASAP2 in OA progression.
Methods: We examined the impact of ASAP2 knockdown or genetic deletion on macrophage polarization, inflammatory responses, and NF-κB signaling. Protein interactions and RNA-binding dynamics were assessed to elucidate the underlying mechanisms. In vivo analyses were conducted using myeloid-specific Asap2 knockout mice in the destabilization of the medial meniscus (DMM) model of OA.
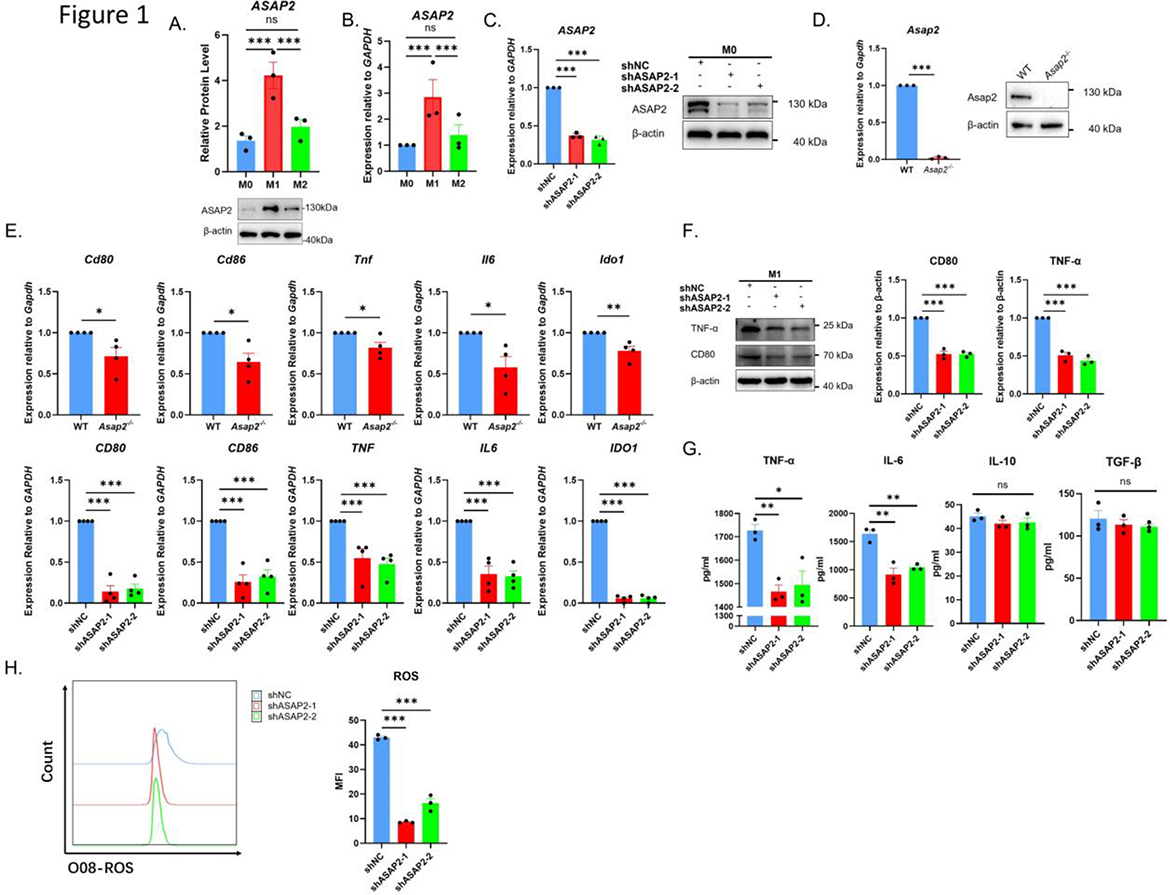
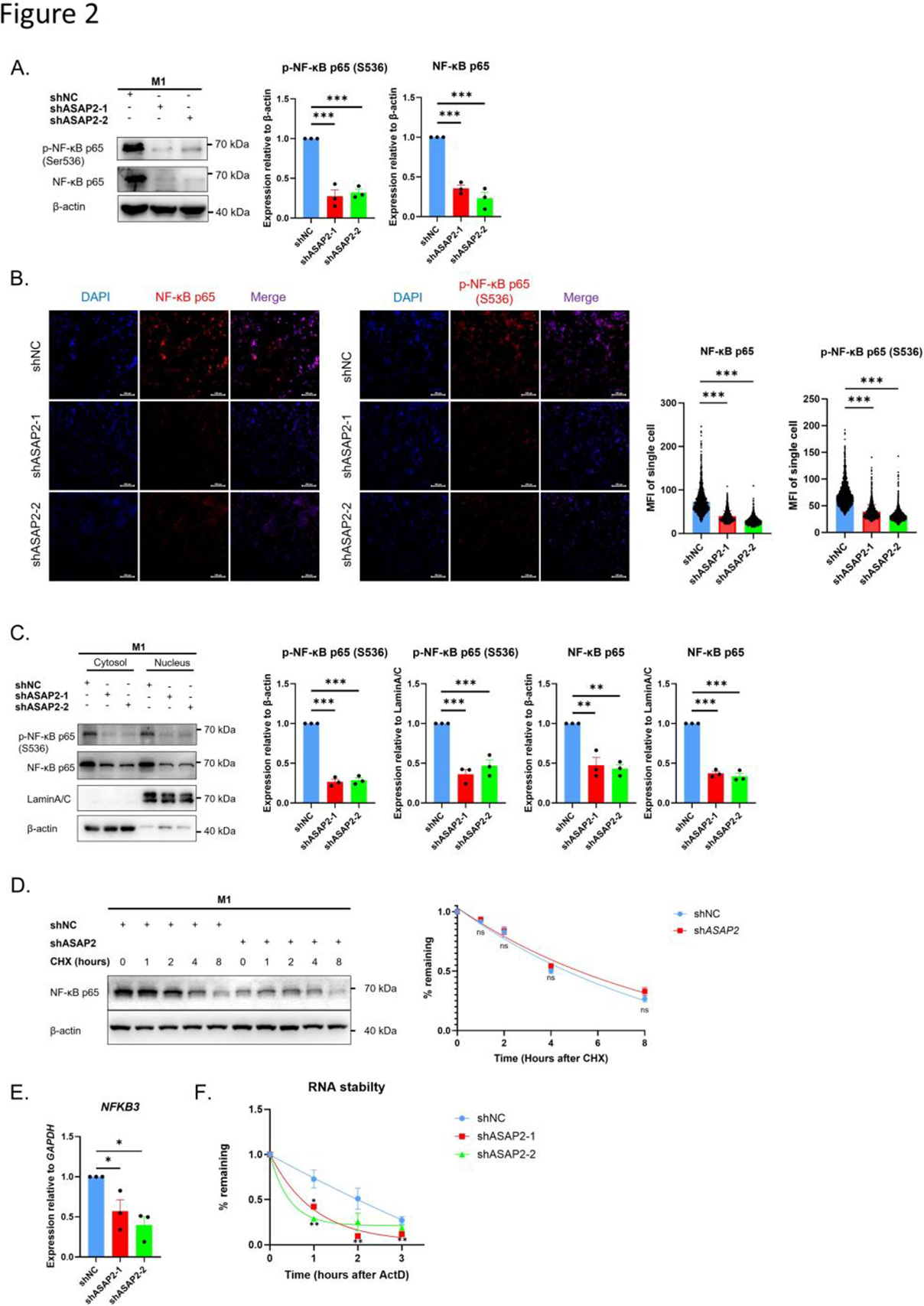
Results: ASAP2 expression was significantly elevated in M1 macrophages (Figure 1). Loss of ASAP2 suppressed M1 polarization, reduced pro-inflammatory cytokine production, and decreased NF-κB p65 levels. Mechanistically, ASAP2 interacted with the RNA-binding protein hnRNPA1 to stabilize NFKB3/RELA mRNA. NF-κB p65, in turn, enhanced ASAP2 transcription, forming a positive feedback loop (Figure 2). In the DMM model, myeloid-specific Asap2 knockout reduced synovial M1 macrophages accumulation and ameliorated OA progression.
Conclusions: ASAP2 promotes macrophage M1 polarization and OA progression by stabilizing NFKB3 mRNA via hnRNPA1. These findings reveal a novel mechanism linking macrophage activation to OA and support ASAP2 as a potential therapeutic target.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.