fetching data ... 
Background: Systemic lupus erythematosus (SLE) is characterised by chronic immune activation, in which CD4 + T cells play a central role in sustaining autoantibody production, cytokine amplification, and tissue inflammation. Increasing evidence indicates that immune cell metabolism is not simply a downstream consequence of activation, but an active determinant of T cell fate, persistence, and pathogenic function. In particular, mitochondrial oxidative phosphorylation (OXPHOS) supports sustained T cell activation and survival; however, excessive or dysregulated mitochondrial activity can promote oxidative stress, impaired energy efficiency, and cellular dysfunction. Mitochondrial content is dynamically regulated during T cell differentiation and activation, with mitochondrial expansion often reflecting increased energetic demand. While this may initially support immune responses, persistent mitochondrial expansion in the setting of chronic inflammation may indicate maladaptive metabolic remodelling, characterised by inefficient energy production and increased reactive oxygen species (ROS) generation. Such bioenergetic stress has been implicated in T cell exhaustion, aberrant cytokine production, and impaired immune regulation. Type I interferons (IFN-I), particularly IFN-α, are key drivers of SLE pathogenesis and exert pleiotropic effects on lymphocyte differentiation, survival, and effector function. IFN-I signalling has also been shown to influence cellular metabolism in a context-dependent manner, either restraining or enhancing bioenergetic activity depending on baseline metabolic state. In SLE, where IFN-I signalling is chronically activated, it remains unclear how IFN-α interacts with intrinsic metabolic abnormalities in CD4 + T cells. Specifically, whether IFN-α exacerbates mitochondrial expansion and bioenergetic dysfunction, thereby sustaining pathogenic T cell activation, has not been clearly defined.
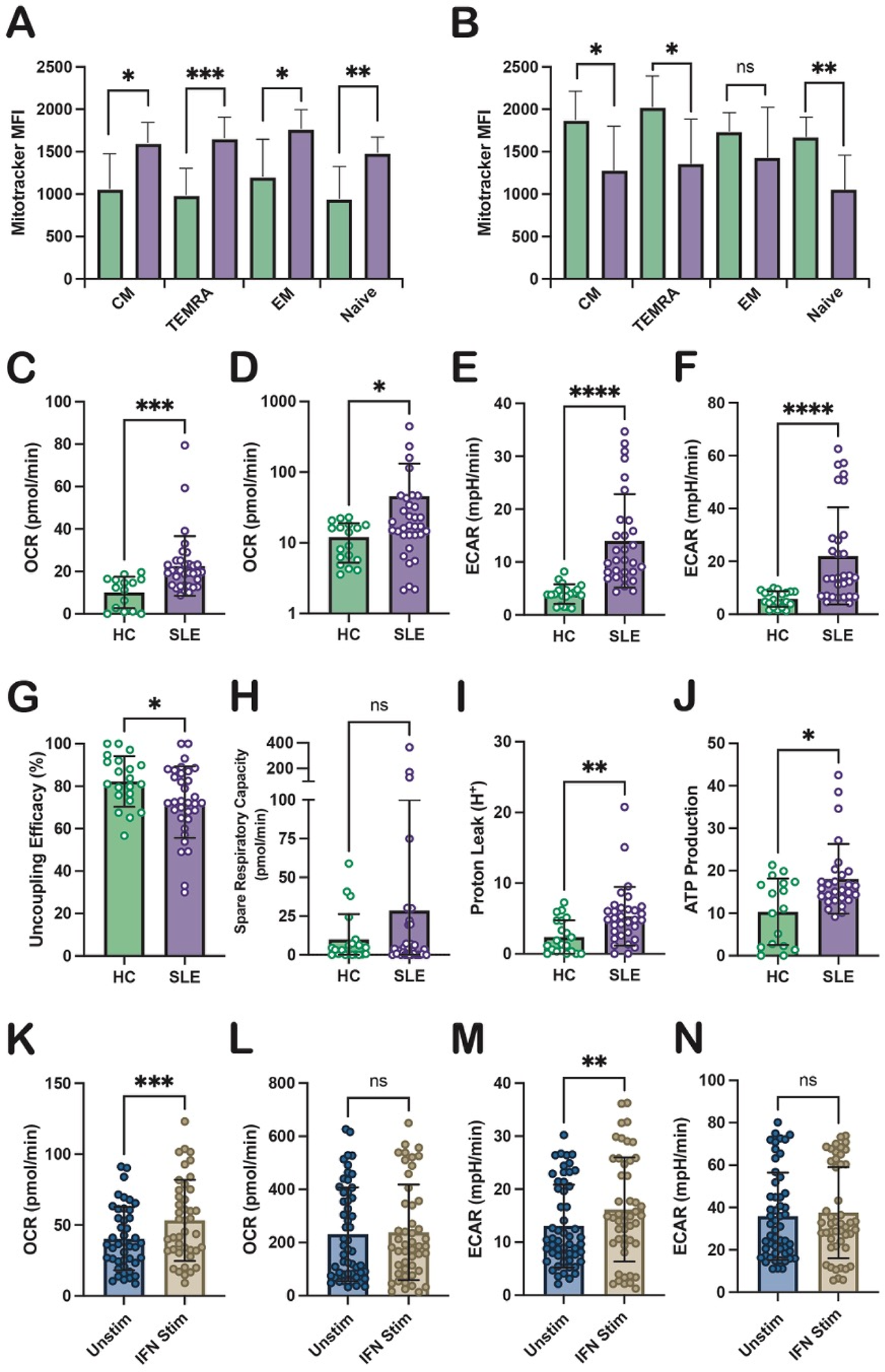
Objectives: To determine whether CD4 + T cells from patients with SLE exhibit altered mitochondrial content and bioenergetic function, and to define how IFN-α modulates these metabolic pathways across CD4 + T cell subsets.
Methods: Patients with well-controlled SLE (n=33; median SLEDAI-2K=2) and healthy controls (HC; n=17) were recruited to minimise confounding effects of overt inflammation or high-dose immunosuppression. Mitochondrial mass was quantified by flow cytometry using MitoTracker staining in CD4 + and CD8 + T cells, including naïve (CD45RO − CCR7 + ), central memory (CM; CD45RO + CCR7 + ), effector memory (EM; CD45RO + CCR7 − ), and TEMRA (CD45RO − CCR7 − ) subsets. CD4 + T cells were negatively enriched from peripheral blood mononuclear cells and profiled using Seahorse XF MitoStress assays to quantify mitochondrial respiration (oxygen consumption rate, OCR) and glycolytic flux (extracellular acidification rate, ECAR). To assess the impact of IFN-I signalling, matched CD4 + T cell aliquots from SLE and HC donors were cultured with or without recombinant human IFN-α (1,000 U/mL, 16 h) prior to bioenergetic profiling.
Results: Baseline mitochondrial content Compared with HC, SLE CD4 + T cells exhibited significantly increased mitochondrial mass (p=0.0064). This expansion was observed across all CD4 + subsets, including CM (p=0.0102), TEMRA (p=0.0008), EM (p=0.0106), and naïve cells (p=0.0049) (Figure 1A), indicating a global remodelling of CD4 + T cell mitochondrial content. In contrast, SLE CD8 + T cells did not demonstrate overall mitochondrial expansion (p=0.416) and instead showed reduced mitochondrial mass within CM (p=0.0261), TEMRA (p=0.0167), and naïve subsets (p=0.0040), with no difference observed in EM cells (p=0.22) (Figure 1B). These findings indicate lineage-specific metabolic adaptation in SLE, with selective vulnerability of CD4 + T cells. Baseline bioenergetics (CD4 + T cells ) Ex vivo SLE CD4 + T cells demonstrated elevated basal (p=0.0002; Figure 1C) and maximal OXPHOS (p=0.0353; Figure 1D), alongside increased basal (p<0.0001; Figure 1E) and maximal glycolysis (p<0.0001; Figure 1F), consistent with a hypermetabolic state. Despite this increased metabolic flux, spare respiratory capacity was unchanged (p=0.84; Figure 1G), suggesting limited ability to respond to additional energetic stress. Importantly, markers of mitochondrial inefficiency were evident, including increased proton leak (p=0.0017; Figure 1H), reduced coupling efficiency (p=0.0249; Figure 1I), and increased ATP production (p=0.0289; Figure 1J). Together, these data indicate mitochondrial expansion accompanied by reduced energetic efficiency and increased bioenergetic stress. IFN-α modulation In HC CD4 + T cells, IFN-α exposure did not significantly alter basal OXPHOS and reduced basal and maximal glycolysis, consistent with IFN-I–mediated metabolic restraint in non-inflamed cells. In contrast, SLE CD4 + T cells displayed heightened sensitivity to IFN-α, with a further increase in basal OXPHOS (p=0.0001; Figure 1K) without changes in maximal OXPHOS (p=0.71; Figure 1L). IFN-α also increased basal glycolytic activity relative to unstimulated SLE cells (p=0.0016; Figure 1M), while maximal ECAR remained unchanged (p=0.57; Figure 1N). These findings suggest that IFN-α amplifies baseline energetic demand in metabolically primed SLE CD4 + T cells, further constraining metabolic reserve.
Conclusions: CD4 + T cells from patients with SLE exhibit marked mitochondrial expansion and a hypermetabolic yet inefficient bioenergetic profile characterised by elevated oxidative and glycolytic flux, limited metabolic flexibility, and features of mitochondrial stress. IFN-α selectively amplifies basal respiratory and glycolytic activity in SLE CD4 + T cells, exacerbating intrinsic metabolic dysfunction while exerting distinct effects in healthy cells. These data support a model in which chronic IFN-driven metabolic rewiring sustains pathogenic CD4 + T cell activation in SLE and highlight bioenergetic pathways as biologically relevant targets for immunometabolic intervention.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: Chris Wincup Biogen, Novartis, UCB, Viatris, AstraZeneca, BMS, CSL Vifor, Johnson & Johnson, Kyverna,Otsuka, Roche, UCB, Abbvie, Gilead, AstraZeneca, George Robinson: None declared, Meredyth Wilkinson: None declared, Anisur Rahman: None declared.