fetching data ... 
Background: Copper (Cu) is an essential micronutrient involved in numerous physiological processes, and its dysregulation is implicated in various diseases, including genetic disorders, cancers, and neurodegenerative conditions. Beyond these, copper plays a critical role in immune function, influencing lymphocyte proliferation, antibody production, and inflammatory responses. Autoimmune diseases (AIDs), such as systemic lupus erythematosus (SLE), are characterized by the immune system attacking host tissues. Given copper’s importance in immune regulation, its potential involvement in AID pathogenesis remains underexplored. While previous studies have reported altered copper levels in SLE patients, a comprehensive understanding of copper metabolic dysregulation—including isotopic, molecular, and functional dimensions—is lacking. Therefore, this study systematically investigates copper metabolism in SLE by integrating phenotypic profiling (concentration and δ 65 Cu), transcriptomic analysis, and in vivo intervention. We aim to clarify whether copper dysregulation is a hallmark of SLE and explore its role in disease mechanisms and progression.
Objectives: Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by immune system attacks on host tissues. Copper is an essential trace element for immune function, yet whether its metabolic dysregulation contributes to SLE pathogenesis remains unclear. This study aimed to systematically investigate alterations in copper metabolism in SLE and its role in disease development.
Methods: Blood samples from 50 patients with autoimmune diseases (mainly SLE) and 34 healthy controls were collected. Copper concentrations and stable isotope ratios (δ 65 Cu) in plasma and blood cells were measured using inductively coupled plasma mass spectrometry (ICP-MS) and multi-collector ICP-MS. Public transcriptomic data from the Gene Expression Omnibus (GEO) database, comprising 264 SLE patients and 83 healthy subjects, were analyzed to assess the expression of copper metabolism-related genes. Gene set enrichment analysis (ssGSEA) was performed to evaluate activity changes in copper metabolism modules. Protein expression was validated by Western blotting. Furthermore, an SLE mouse model (MRL/lpr) was used to evaluate the effects of modulating copper metabolism with the copper chelator ammonium tetrathiomolybdate (TTM) on disease progression.
Results: Compared with healthy controls, SLE patients showed significantly elevated plasma copper concentration ( P < 0.01) and decreased δ 65 Cu in both plasma and blood cells ( P < 0.0001). Transcriptomic analysis revealed dysregulation of multiple copper metabolism-related genes in SLE patients, including those involved in copper uptake (e.g., upregulated STEAP4 and SLC31A1 ), efflux (e.g., upregulated ATP7A and ATP7B ), mitochondrial respiration, and cuproptosis. At the protein level, SLE patients showed increased ceruloplasmin (Cp) and decreased albumin (Alb) in plasma, along with elevated STEAP4 protein in blood cells. Similar copper metabolism abnormalities were observed in MRL/lpr mice. Intervention with the copper chelator TTM significantly alleviated kidney inflammation, fibrosis, lymphadenopathy, and immune dysfunction in the mouse model, and partially reversed disease-associated gene expression changes.
Conclusions: This study systematically elucidates significant dysregulation of copper metabolism in SLE. Aberrant copper metabolism likely contributes to SLE pathogenesis through pathways involving mitochondrial respiration and collagen cross-linking. Copper chelation modulated disease progression in an SLE mouse model, offering novel insights into the mechanisms of SLE and potential therapeutic strategies.
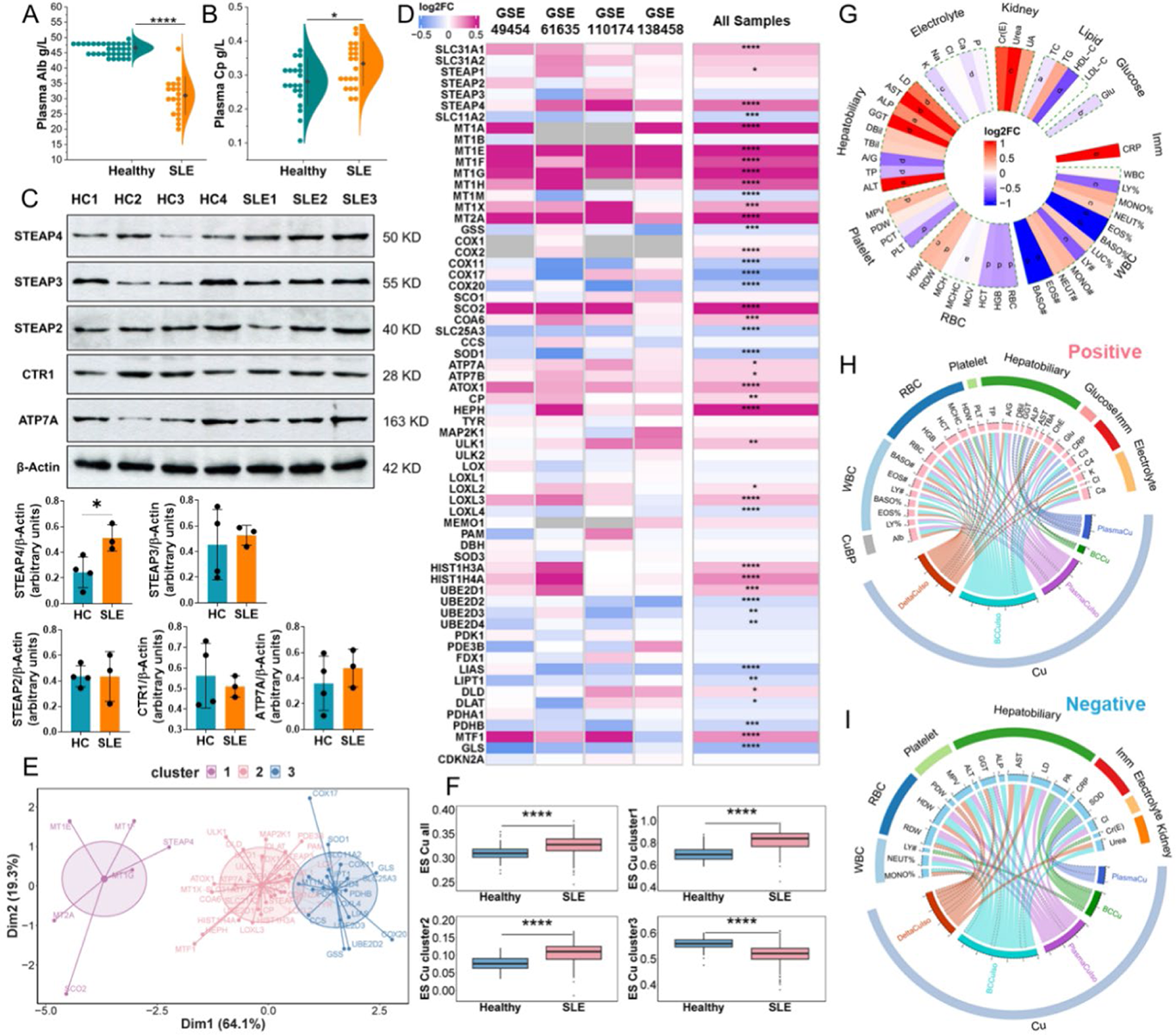
Alterations in molecules within Cu metabolism and correlations between Cu metabolism and clinical parameters in SLE. (A and B) Alb and Cp protein concentration in the plasma. (C) The expression of STEAP2, STEAP3, STEAP4, CTR1, and ATP7A proteins in the BC samples. (D) The transcriptional fold change (FC, SLE-PS vs healthy-PS) of genes in the Cu module. (E) The clustering result of genes in the Cu module. (F) ES of genes in the Cu module (-all and cluster 1, 2, 3) calculated by ssGSEA algorithm. 0.01 ≤ P < 0.05 *, 0.001 ≤ P < 0.01 **, 0.0001≤ P < 0.001 ***, P < 0.0001 ****. (G) Differential analysis of clinical parameters between SLE-TS and healthy-TS.
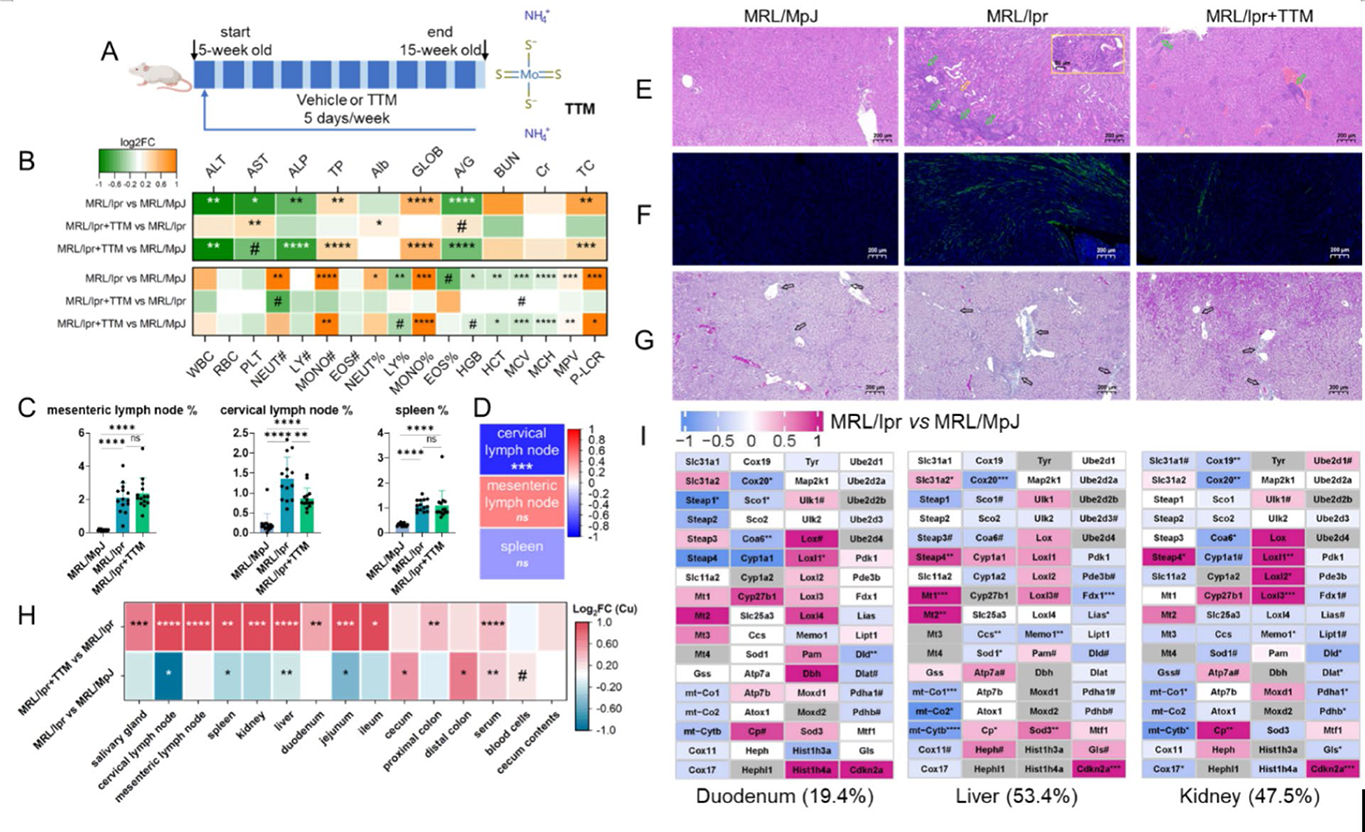
Analysis of biological features and Cu metabolism in SLE mouse model. (A) The mouse experimental design. (B) Log 2 FC of physiological indexes in the mice. The abbreviations are explained in Table S5. (C) Relative weight of mouse immune organs ( n = 14 in each group). (D) Spearman correlation between Cu concentration and relative weight of mouse immune organs. The data from the MRL/lpr+TTM group are excluded because TTM reduced the bioavailability of Cu. (E–G) Representative images of H&E staining, immunofluorescence staining of IgG (green fluorescence), and Masson’s trichrome staining in mouse kidney sections. In (E), the green arrows point at the areas with relative severe inflammation, and the yellow arrow point at a representative crescentic structure and its magnified image is in the yellow box. In (G), the blue areas indicated by the black arrow exist deposition of collagen fibers. (H) Log 2 FC of Cu concentrations in multiple tissues of mice. (I) Transcriptional log 2 FC of genes in mouse Cu module.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.