fetching data ... 
Background: Systemic lupus erythematosus-associated immune thrombocytopenia (SLE-ITP) develops under persistent immune dysregulation and is associated with an adverse prognosis. Moreover, a subset of patients remains refractory to standard immunosuppression therapies and B-cell-targeting strategies. The heterogeneity in clinical characteristics and therapeutic responses reflects a complex pathogenesis that cannot be fully explained by peripheral immune-mediated platelet destruction, suggesting an underappreciated contribution of reduced bone marrow (BM) megakaryopoiesis. However, the molecular mechanisms underlying impaired megakaryopoiesis in SLE-ITP remain poorly defined, thus hindering the development of effective therapies.
Objectives: This study aims to characterize BM megakaryopoietic defects in SLE-ITP, evaluate GATA1-HES1 dysregulation in hematopoietic stem and progenitor cells (HSPCs) under sustained immune activation, which impairs megakaryocyte (MK) production, and assess whether the histone deacetylase inhibitor chidamide (CHI) can reprogram these pathways to restore megakaryopoiesis.
Methods: The morphology of BM was assessed in 28 SLE-ITP patients. MK abundance and protein levels were quantified by flow cytometry in BM samples from SLE-ITP (n=4), non-ITP SLE (n=3), and healthy controls (HC, n=4). SLE immune-stress conditions were modeled ex vivo using the TLR7 agonist R848 in mice whole BM and Lin - HSPC cultures. Bulk RNA sequencing was performed in HSPCs to identify shared, differentially expressed pathways. CHI effects were evaluated in patient BM cultures and in vivo in mice. CUT&Tag- sequencing was used to assess epigenetic changes, while HES1 protein stability was evaluated via CHX chase assay combined with MG132. Finally, the dependency on GATA1 was tested by siRNA knockdown.
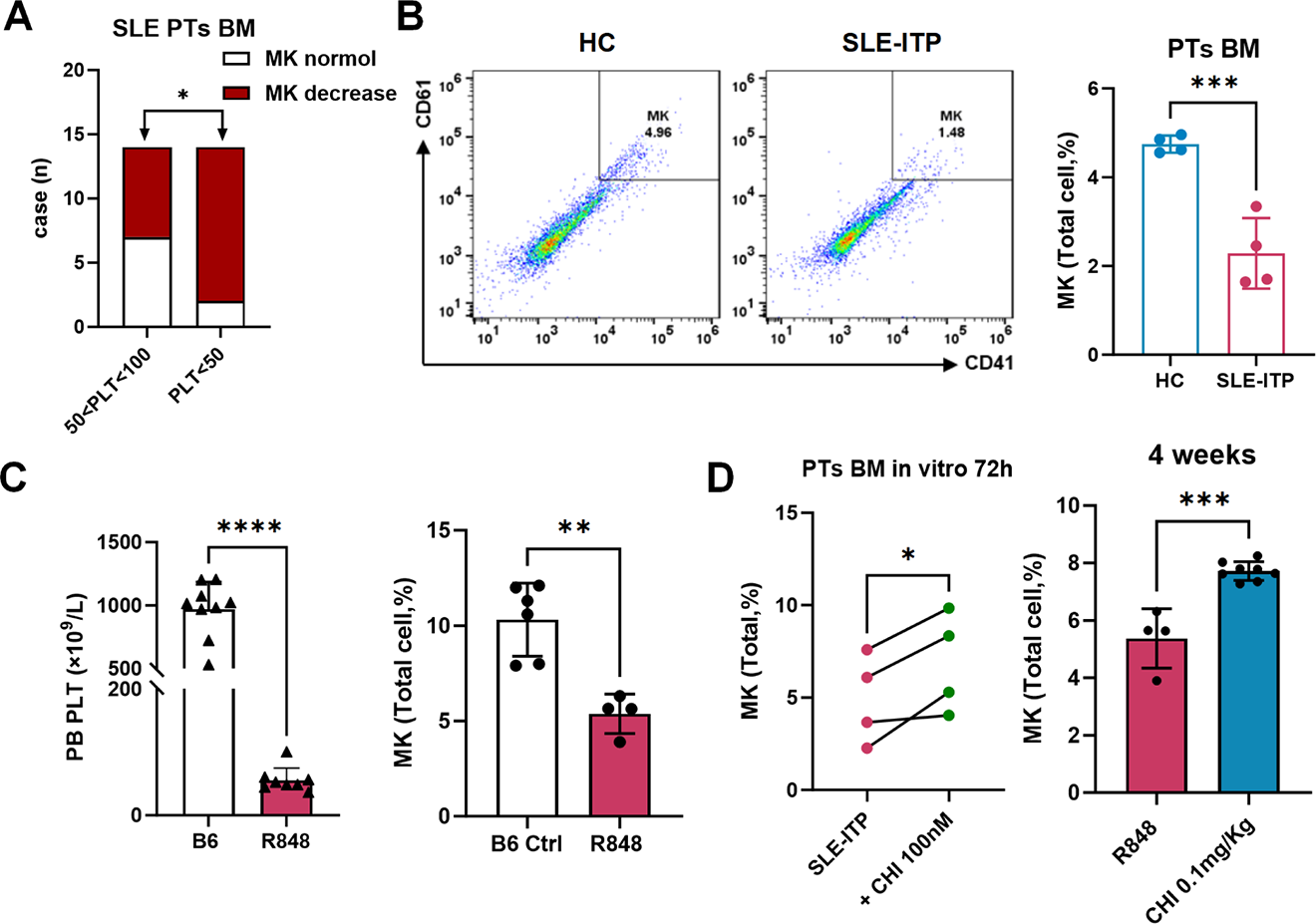
Results: In our cohort, SLE-ITP patients with reduced BM MK proportions exhibited significantly lower peripheral platelet counts, indicating impaired megakaryopoiesis as a more severe disease phenotype (Figure 1A). A marked reduction in the proportion of MKs was observed in the BM of SLE-ITP patients compared to HC, a consistent feature also present in the murine model (Figure 1B-C), supporting a disease-related defect in platelet production. Moreover, R848 suppressed MK output in vitro in whole BM and purified Lin − HSPCs, respectively, indicating that immune-inflammatory stimulation can constrain megakaryopoietic programs at the progenitor level. Transcriptomic profiling of patient-derived CD34 + HSPCs and murine Lin − HSPCs converged to identify HES1 as a key dysregulated gene, with enrichment of platelet-related pathways and transcriptional networks associated with GATA1 and HDAC1. Therefore, we measured the acetylation level and found CD34 + HSPCs from SLE-ITP patients exhibited low H3K27 acetylation (H3K27ac) and GATA1 expression, accompanied by aberrant accumulation of transcriptional repressor HES1, compared with HC and non-ITP SLE samples; a similar pattern was observed in the murine model. CHI treatment promoted MK output in patient BM samples and mouse models (Figure 1D). It also upregulated MK lineage markers, including Itga2b, Itgb3, and Gp1bb, and increased platelet levels in mice. Furthermore, CHI increased patient CD34 + HSPCs H3K27ac and GATA1 protein levels, while downregulating HES1 protein levels. Mechanistically, in patient BM samples, epigenomics data demonstrated that CHI enhanced H3K27ac enrichment at the GATA1 promoter and increased GATA1 occupancy at the promoters of MK lineage markers, as well as the HES1 promoter. In parallel, CHI enhanced HES1 proteasomal degradation and weakened HES1 binding to the MK gene promoters and its own promoter. The knockdown of GATA1 partially withdrew CHI-induced MK recovery, implying a GATA1-dependent reprogramming mechanism. (Figure 2).
Conclusions: Our study suggested that epigenetic and proteostatic dysregulation of the GATA1-HES1 pathway in HSPCs under immune-inflammatory stress is associated with impaired megakaryopoiesis and reduced platelet output in SLE-ITP. Reprogramming of this pathway by CHI restored MK output through coordinated chromatin activation and protein homeostasis, supporting BM defect in megakaryopoiesis and implying a potential therapeutic strategy for SLE-ITP.
(A) Statistical analysis of the bone marrow MK count and peripheral platelet count in SLE-ITP. (B) Bone marrow MK proportion in SLE-ITP patients versus healthy controls. (C) Bone marrow MK proportion and peripheral platelet count in the SLE-ITP mouse model compared to the B6 control. (D) MK proportion after chidamide treatment in SLE-ITP patient bone marrow samples and mouse models.
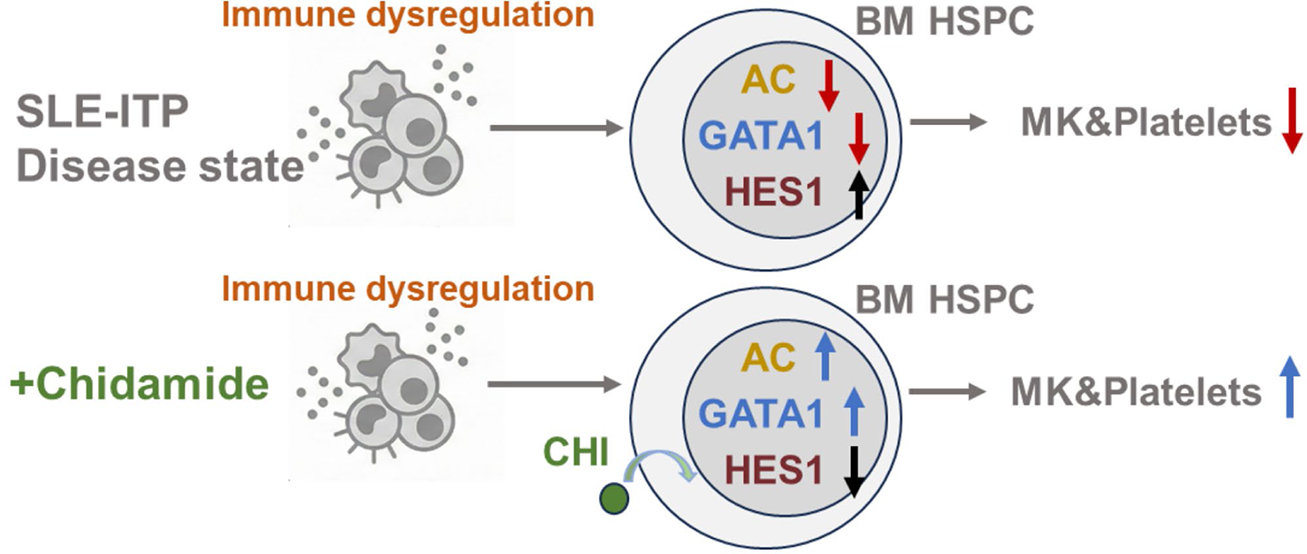
Graphical abstract. Under immune stress, epigenetic suppression of GATA1 and protein accumulation of HES1 in SLE-ITP bone marrow HSPCs, constraining megakaryopoiesis and platelet production, a condition that is reversible with chidamide.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.