fetching data ... 
Background: Behçet’s syndrome (BS) is a chronic multisystemic disease whose manifestations include mainly oral and genital ulcerations, uveitis, central nervous system disorders, enterocolitis and vascular involvement [1]. Although its etiology remains unexplained, the microbiota could play a triggering role in BS [2]. Previously published studies suggest that BS patients have an altered gut microbiota composition that could impact their inflammatory and immune landscape [2,3]. However, all BS microbiota studies have obvious limitations as the results have not been adjusted for treatment and environmental factors such as tobacco consumption or diet.
Objectives: The aim of this study was to compare and correlate the following parameters in BS patients and matched healthy controls (HC) living in the same environment and sharing the same food dietary, according to clinical phenotypes and treatments: gut microbiota, serum markers of inflammation, serum and salivary cytokines concentrations (IL-1β, IL-17A, IL-18, IFN-γ, TNF-α, IL-6 and IL-22), circulating amino acids concentrations, Short Chain Fatty Acids (SCFA) concentrations in stools.
Methods: BEHCETBIOT is a French multicenter case-control study that included patients meeting the international classification criteria for Behçet’s disease revised in 2013 [4], who developed the disease within 10 years before the study. HC were recruited from the patients’ environment. Stool, blood and saliva samples were taken from the entire study population. Following stool DNA extraction, 16S rRNA gene was amplified and sequenced (V3-V4 regions, Qiagen, USA). MicrobiomeStat R package was used to analyze 16s rRNA sequences. For the taxonomic analysis, features were assigned to operational taxonomic units (OTU) with MTP pipeline (Microbiome Taxonomic Profiling) referred to ezBiocloud database. A linear regression framework (LinDA) was performed for differential abundance analysis (FDR-adjusted p-value < 0.1). To evaluate the discriminatory potential of gut microbiota composition for BS diagnosis, sparse Partial Least Squares Discriminant Analysis (sPLS-DA) was applied at genus level. IL-1β, IL-17A, IL-18, IFN-γ, TNF-α, IL-6 and IL-22 concentrations were measured in serum and saliva by Ella® automated ELISA (Bio-Techne). Amino acids concentrations in serum as well as SCFA concentrations in stools were measured by Liquid Chromatography Mass Spectrometry. Paired univariate comparisons were performed between BS cases (or subgroups of BS cases) and HC to assess fold change, standardized effect size (Cohen’s d) and statistical significance (Welch’s t-test). Multivariate analyses including all the parameters studied were performed using R Core Team (2023) to obtain co-expression networks.
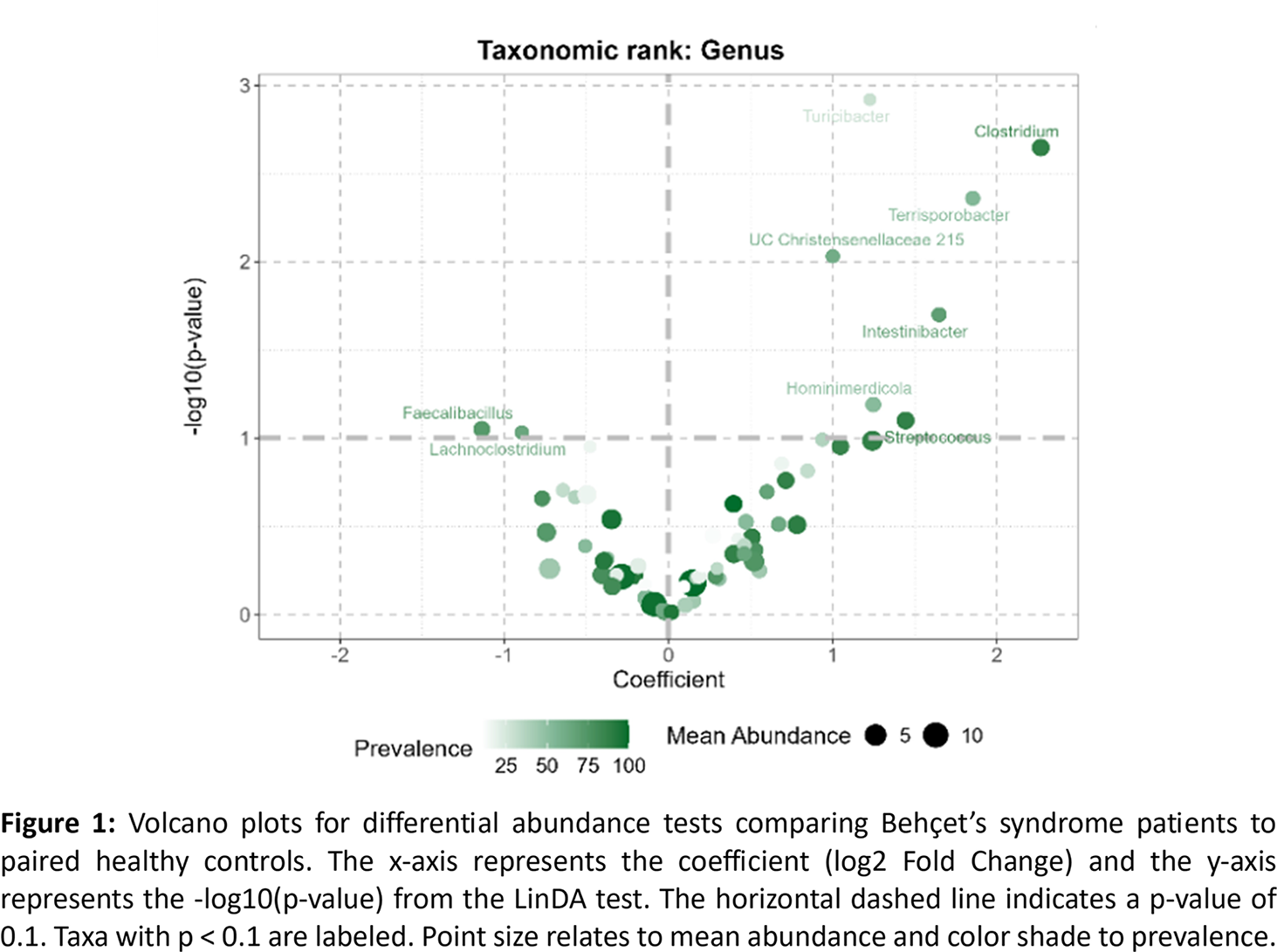
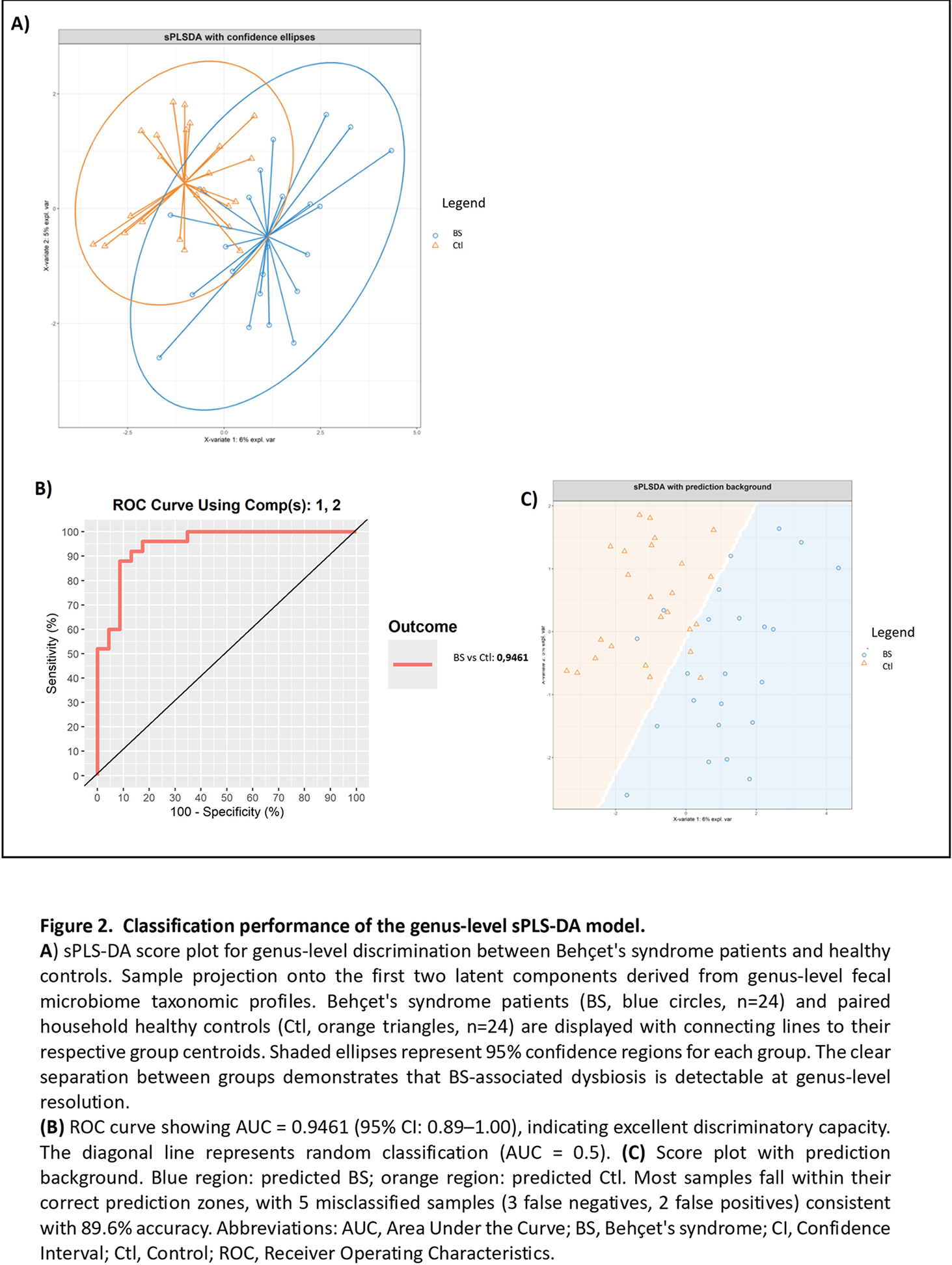
Results: Thirty-three BS patients (18 women – 54,5 %) and paired HC were enrolled. Main BS clinical profiles included mucocutaneous and articular phenotype (33%), neurological and/or ocular phenotype (42%) and vascular phenotype (24%). All patients had inactive BS at inclusion. Treatment included anti-TNF-α in 27%, anti-IL-6 in 9%, and colchicine in 81% at the time of inclusion. Differential abundance analysis using linear discriminant analysis identified significant taxonomic shifts between BS patients and their paired HC. A total of 47 taxa showed differential abundances across all taxonomic ranks. Bacterial families Clostridiaceae, Peptostreptococcaceae and Turicibacteraceae were significantly more abundant in BS patients than HC. This was confirmed at the genus level by a significant over-representation of Clostridium , Turicibacter , Terrisporobacter , Intestinibacter and unclassified Christensenellaceae, as well as a trend (FDR < 0.1) for enrichment of Streptococcus and Hominimerdicola ( Figure 1 ). Interestingly, the three BS clinical phenotypes showed distinct microbiota features, the neuro-ophtalmic phenotype being associated with a loss of Desulfobacteriota whereas the vascular phenotype displayed massive Enterobacteriaceae depletion. sPLS-DA shows that fecal microbiota composition at genus level can accurately discriminate BS patients and matched HCs ( Figure 2A ). Classification performance was excellent (89.6% accuracy, Kappa 0.792, AUC 0.9461, Figure 2BetC ) with 11 discriminant genera enriched in HC ( Lachnoclostridium , Faecalibacillus , Roseburia ) or in BS ( Streptococcus , Clostridium , Turicibacter ). Regarding cytokines, microbiome metabolites and inflammatory parameters, co-expression integrating network highlights a pro-inflammatory signature in BS and shows a distinct SCFA cluster linked to inflammation parameters, reinforcing the hypothesis of a common microbiota-immune contribution to BS physiopathology.
Conclusions: This paired case-control study reveals profound and heterogeneous gut microbiome alterations in BS patients. Of note, the enrollment of matched HC sharing the same environment strengthens our findings by minimizing lifestyle-driven differences. Despite modest sample sizes and substantial inter-individual variability, distinct microbial signatures were associated with specific clinical phenotypes, suggesting that distinct microbial communities could contribute to clinical heterogeneity. Functional studies will be required to understand how these microbial alterations could lead to pathophysiological consequences. Further work will determine whether a BS-specific signature could identify among patients with idiopathic recurrent aphthous ulcerations those who are likely to develop true Behçet’s syndrome.
REFERENCES: [1] Saadoun D et al. Behçet’s Syndrome. N Engl J Med 2024;390:640–51.
[2] Joubert M, et al. Microbiome and Behçet’s disease: a systematic review. Clin Exp Rheumatol 2023;41:2093–104.
[3] Consolandi C, et al. Behçet’s syndrome patients exhibit specific microbiome signature. Autoimmunity Reviews 2015;14:269–76.
Acknowledgments: NIL.
Disclosure of Interests: None declared.