fetching data ... 
Background: Systemic lupus erythematosus (SLE) is an autoimmune disease characterised by excessive differentiation of B cells into plasmablasts, with disease pathogenesis shaped by both genetic and environmental factors. As genetic factors, evidence from genome-wide association studies and studies of inborn errors of immunity has identified abnormalities in type I interferon signalling and B-cell differentiation involving genes such as TLR7, TNFAIP3 , and IRF5 . In addition to environmental triggers such as ultraviolet exposure and sex hormones, the gut microbiota has emerged as an important factor to SLE pathogenesis. Recent studies have demonstrated reduced gut microbial diversity (dysbiosis) in lupus patients, characterised by a decrease in short-chain fatty acid (SCFA)–producing Faecalibacterium species and an increase in pro-inflammatory Streptococcus species. Gut dysbiosis affects intestinal immunity and alters gut microbiota-derived metabolites; however, the impact of these metabolites on immune cell differentiation and disease pathophysiology remains unclear
Objectives: This study investigated the effect of gut microbiota–derived metabolites on B-cell differentiation, particularly plasmablast differentiation, and their role in the pathogenesis of SLE.
Methods: CD19 + B cells isolated from peripheral blood of healthy controls and lupus patients were cultured in the presence of various gut microbiota–derived metabolites, including SCFAs, secondary bile acids, tryptophan metabolites, desaminotyrosine, and trimethylamine N-oxide. Plasmablast differentiation was assessed by flow cytometry. The expression of transcription factors involved in plasmablast differentiation was evaluated by quantitative PCR, and immunoglobulin production was measured by ELISA. To elucidate the underlying mechanisms of function of these metabolites, metabolic flux analysis, Western blotting, and chromatin immunoprecipitation (ChIP) assays were performed. In addition, serum concentrations of gut microbiota-derived metabolites in healthy controls and lupus patients were quantified using high-performance liquid chromatography (HPLC). This study was approved by the ethics committee of the University of Occupational and Environmental Health, Japan (approval numbers: #UOEHCRB19-046 and #UOEHCRB21-069).
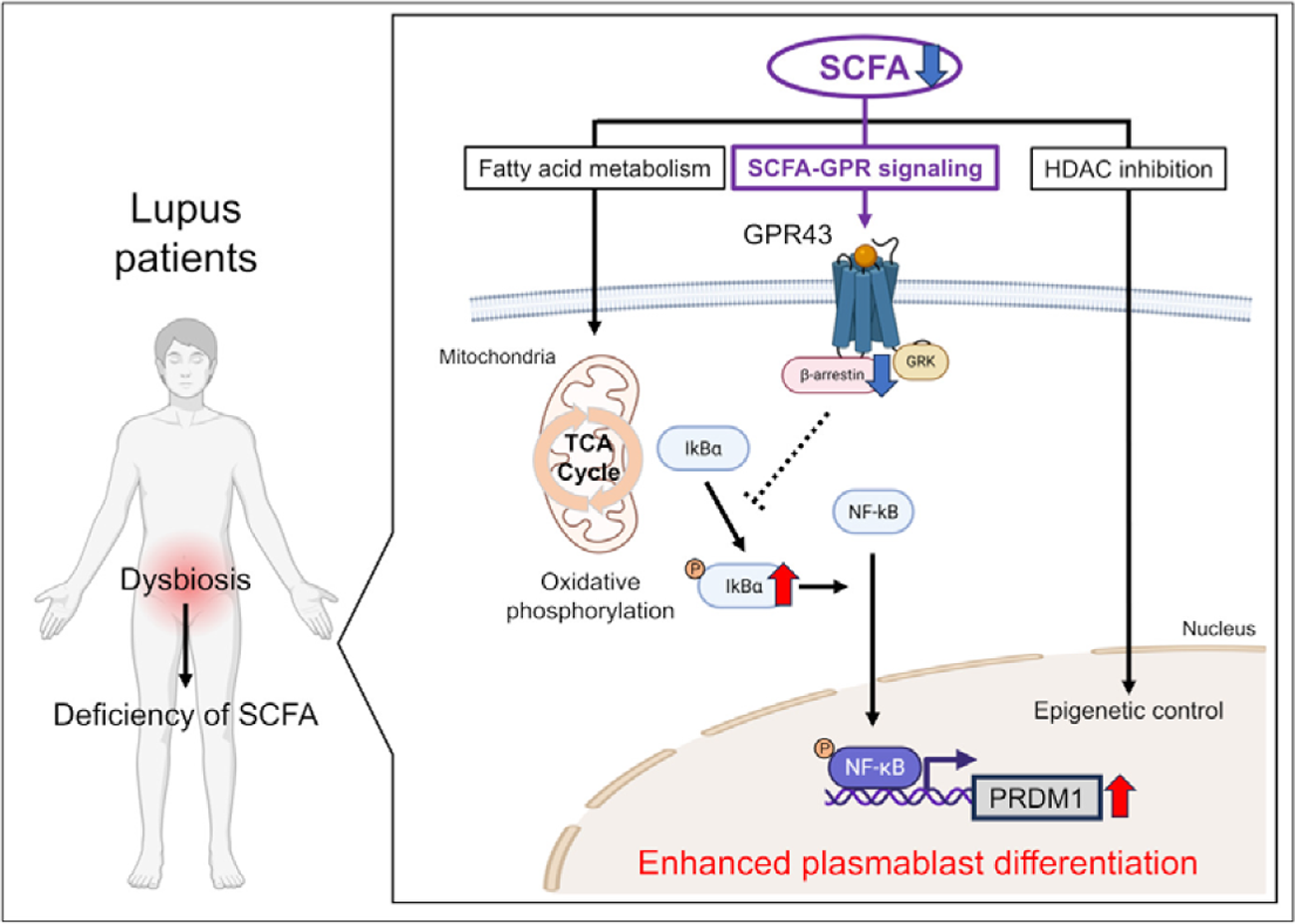
Results: In vitro, CD19 + B cells robustly differentiated into plasmablasts upon stimulation with B-cell receptor cross-linking, CpG (TLR ligand), and interferon-α. Most gut microbiota–derived metabolites did not affect plasmablast differentiation under these conditions. However, the addition of SCFAs (butyrate, propionate, and acetate) suppressed plasmablast differentiation in a dose-dependent manner, leading to reduced immunoglobulin production and decreased expression of the differentiation-promoting factor, PR domain zinc finger protein 1 ( PRDM1 ). SCFAs exert their biological effects through (i) fatty acid metabolism, (ii) histone deacetylase (HDAC) inhibition, and (iii) G protein–coupled receptor (GPR) signalling. Metabolic flux analysis demonstrated that SCFAs enhanced oxidative phosphorylation in B cells, an effect reversed by the fatty acid oxidation inhibitor etomoxir. However, inhibition of fatty acid metabolism by etomoxir did not reverse SCFA-induced suppression of differentiation. Regarding HDAC inhibition, the HDAC inhibitor trichostatin A suppressed plasmablast differentiation when HDAC inhibition exceeded 30%. In contrast, HDAC inhibitory activity of SCFAs was relatively weak, reaching 8% for acetate and up to 22% for butyrate, suggesting a limited contribution of HDAC inhibition to the suppression of differentiation. With respect to GPR signalling, human B cells expressed the SCFA receptors GPR41 and GPR43. Activation of GPR43 by agonist, but not GPR41, suppressed plasmablast differentiation. Moreover, the inhibitory effect of SCFAs was reversed in the presence of a GPR43 antagonist. SCFA stimulation or GPR43 signalling induced activation of intracellular β-arrestin 2. Downstream of β-arrestin 2, phosphorylation of IκBα and NF-κB was suppressed, resulting in reduced NF-κB binding to the PRDM1 promoter. These findings indicate that SCFAs suppress plasmablast differentiation via the GPR43–β-arrestin 2 signalling pathway. Finally, serum SCFA concentration was significantly reduced in lupus patients (n=35) compared with healthy controls (n=18). Addition of SCFAs or a GPR43 agonist to peripheral blood B cells from lupus patients suppressed plasmablast differentiation and reduced immunoglobulin production.
Conclusions: Deficiency of serum SCFA contributed to aberrant plasmablast differentiation in SLE, mediated predominantly through SCFA-specific GPR43 signalling. Fine-tuning of GPR43-mediated signalling may provide therapeutic insight for SLE. Gut dysbiosis has been reported in patients with systemic lupus erythematosus (SLE) and we revealed that serum levels of short-chain fatty acid (SCFA) were reduced in those with high disease activity. Notably, SCFAs suppressed plasmablast differentiation of human B cells derived from lupus patients. SCFAs exert their biological effects through (i) fatty acid metabolism, (ii) histone deacetylase (HDAC) inhibition, and (iii) G protein–coupled receptor (GPR) signalling. The metabolic and HDAC-inhibitory effects of SCFAs showed limited influence on B cell differentiation. In contrast, signalling through GPR43, a receptor specific for SCFAs, mediated the effects of SCFAs on B cell differentiation. Activation of GPR43 suppressed B cell differentiation, and SCFA stimulation or GPR43 signalling induced activation of intracellular β-arrestin 2. Moreover, phosphorylation of IκBα and NF-κB was suppressed, resulting in reduced NF-κB binding to the PR domain zinc finger protein 1 ( PRDM1 ) promoter. These findings suggest that reduced serum SCFA levels and impaired SCFA-GPR43 signalling in lupus patients may contribute to enhanced plasmablast differentiation. Fine-tuning of GPR43-mediated signalling may provide therapeutic insight for SLE.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: Ryuichiro Kanda R. Kanda has received honoraria for lectures from AbbVie, Chugai, and Taisho., Satoshi Kubo S. Kubo has received speaking fees from Eli Lilly, Bristol-Myers, GlaxoSmithKline, Abbvie, and also research grants from Daiichi-Sankyo, Abbvie, Boehringer Ingelheim, and Astellas., Kaoru Yamagata: None declared, Shigeru Iwata S. Iwata has received honoraria from AbbVie, Asahi- Kasei, Astellas, AstraZeneca, Chugai, Eisai, Daiichi Sankyo, GlaxoSmithKline, Kissei Pharmaceutical, Taisho Toyama Pharmaceutical, UCB Japan, Viatris Pharmaceuticals, Otsuka Pharmaceutical, Eli Lilly, Janssen, Mitsubishi-Tanabe, Boehringer Ingelheim, Novartis Pharma K.K., and Pfizer., Yasuyuki Todoroki: None declared, Masanobu Ueno M. Ueno has received speaking fee from GlaxoSmithKline, Taisho, AstraZeneca, and UCB Japan., Atsushi NAGAYASU: None declared, Yurie Satoh-Kanda Y. Satoh-Kanda has received speaking fees and/or honoraria from Bristol-Myers, Astellas, Boehringer Ingelheim, Taisho, and UCB Japan., Koshiro Sonomoto K. Sonomoto has received speaking fee from Abbvie, Eli Lilly, Gilead Sciences, GlaxoSmithKline, Janssen, Pfizer, UCB Japan, Astellas, Ayumi, Chugai, Taisho, Mitsubishi-Tanabe, and has received research funding from UCB Japan., Ippei Miyagawa I. Miyagawa has received speaking fees from UCB Japan, Eli Lilly, Novartis Pharma K.K., and Abbvie., Yoshiya Tanaka Y. Tanaka has received speaking fees and/or honoraria from Gilead Sciences, AbbVie, Boehringer Ingelheim, Eli Lilly, Mitsubishi-Tanabe, Chugai, Amgen, YL Biologics, Eisai, Astellas, Bristol-Myers, and AstraZeneca; received research grants from Asahi-Kasei, AbbVie, Chugai, Mitsubishi-Tanabe, Eisai, Takeda, Corrona, Daiichi-Sankyo, Kowa, and Boehringer Ingelheim; and received consultant fees from Eli Lilly, Daiichi-Sankyo, Taisho, Ayumi, Sanofi, GlaxoSmithKline, and AbbVie., Shingo Nakayamada S. Nakayamada has received speaking fees and/or honoraria from GlaxoSmithKline, Astellas, Eli Lilly, AbbVie, Mitsubishi-Tanabe, Janssen, Pfizer, Bristol-Myers, Asahi-Kasei, Chugai, AstraZeneca, Taisho, Eisai, Gilead Sciences, UCB Japan, and Ayumi; and has received consulting fees and lecture fees from Asahi-Kasei, Otsuka Pharmaceutical, and Mitsubishi-Tanabe.