fetching data ... 
Background: Targeted B cell depletion via autologous CD19 Chimeric Antigen Receptor T cell (CAR-T) has been shown to induce durable drug-free remission in SLE through deep depletion of B cells in the tissues [1]. However, autologous CD19 CAR-T cell therapy faces limitations constrained by patient-specific manufacturing, high costs, and the need to pause immunosuppressive therapies around leukapheresis which can present health risks. In a first-in-human study, an allogeneic, healthy-donor-derived CD19-targeting synthetic TCR and antigen receptor (STAR)-T cell product induced clinical remission in five patients with severe, refractory SLE complicated with lupus nephritis (LN) [2]. Beyond clinical remission, defining molecular remission induced by CAR-T cell therapies remains challenging, given that numerous gene expression signatures for SLE have been developed. Additionally, it is unclear whether autologous and allogeneic CD19 CAR-T therapies induce similar transcriptional resets when comparing existing SLE signatures
Objectives: To A) systematically compare the overlap of published gene expression signatures in SLE, B) describe how CAR-T cell therapies impact gene expression in longitudinal analyses, and C) compare if and how molecular responses to autologous and allogeneic CAR-T cell therapies differ on a transcriptomic level.
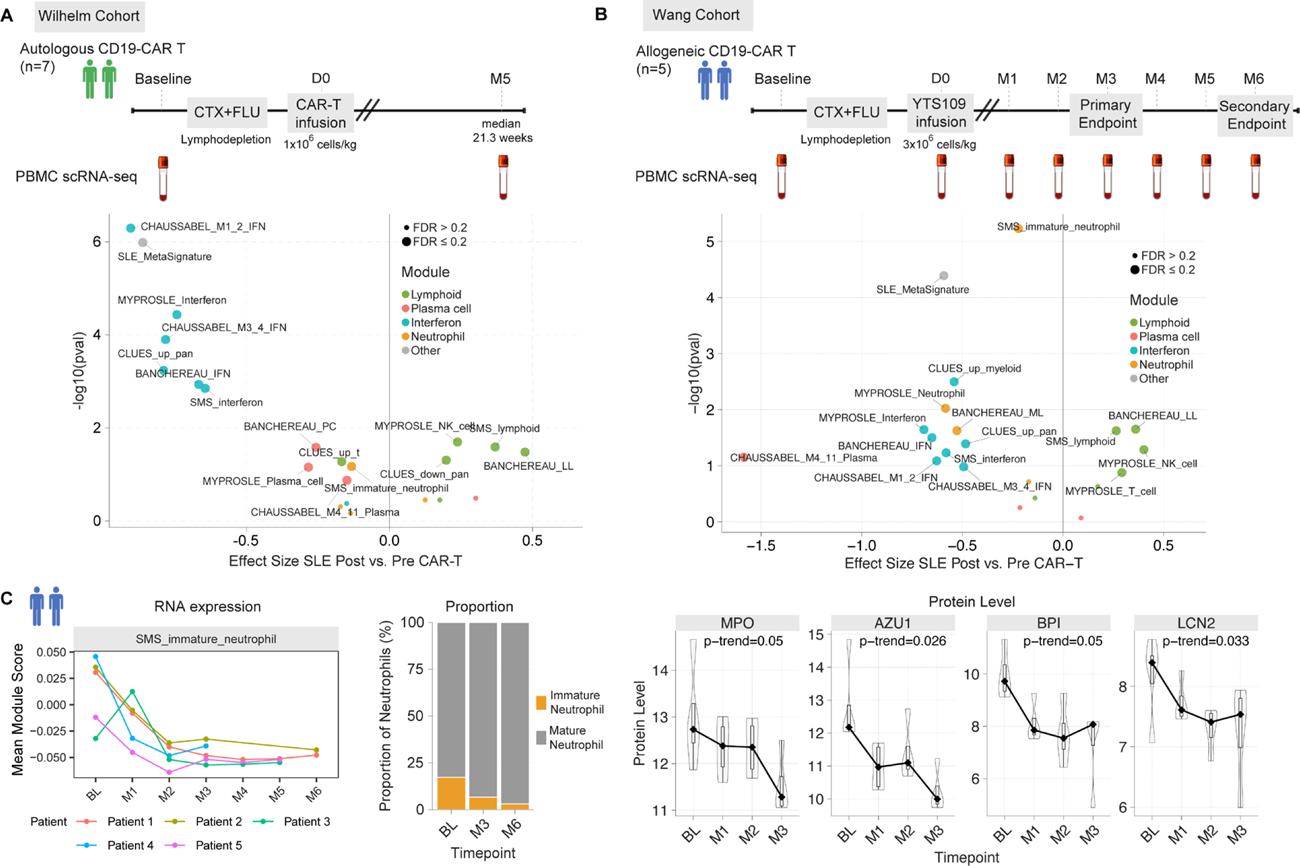
Methods: We analyzed 22 bulk transcriptome gene expression datasets from Gene Expression Omnibus consisting of 15 whole blood and 7 PBMC datasets with 3,797 SLE patients and 841 healthy controls. Using geometric mean expression, we applied previously defined SLE gene expression signatures: Chaussabel, Banchereau, MyPROSLE, CLUES cohort (Perez et al., 2022), and the SLE MetaSignature (SMS). Next, we used single-cell transcriptomics data from two published CD19-CAR T cell therapy cohorts, performing paired differential expression analysis using single-cell MetaIntegrator to evaluate the effects of CAR-T therapy on each of the SLE transcriptional modules. The Wilhelm cohort consists of 7 treatment-refractory SLE patients treated with autologous CD19 CAR-T cell therapy at University Clinic Erlangen [3]. Patients received a single infusion of autologous CD19-targeted CAR T cells after lymphodepletion with fludarabine and cyclophosphamide. The pre-treatment sequencing timepoint was performed prior to lymphodepletion and the post CAR-T therapy timepoint was selected when B cells had repopulated de novo in the peripheral blood following the initial depletion (median time of 21.3 weeks from infusion). The Wang cohort (NCT06379646) consists of 5 treatment-refractory SLE patients complicated by LN treated with an allogeneic T cell product (YTS109) with a CD19-targeting synthetic TCR and antigen receptor (STAR) at Shanghai Changzheng Hospital [2]. All patients received lymphodepletion with fludarabine and cyclophosphamide, followed by a single allogeneic YTS109 infusion on day 0. Allogeneic YTS109 CD19-STAR T cells were manufactured from healthy donor T cells to generate a hypoimmunogenic, off-the-shelf product. PBMCs of trial participants were collected for single-cell sequencing prior to lymphodepletion (baseline), day 0 (YTS10 infusion), day 14, M1, M2, M3, M4, M5 and M6.
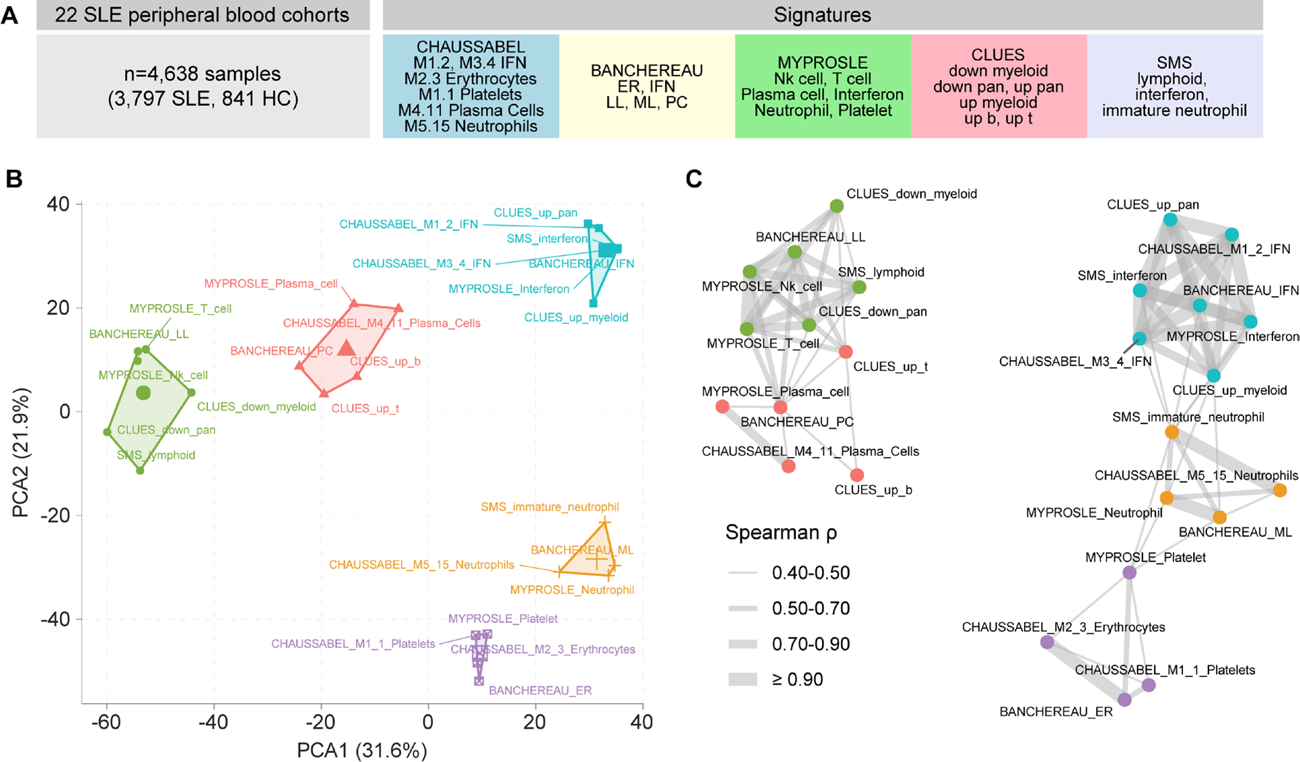
Results: We performed principal component analysis, and network analysis on 3,797 SLE samples to assess signature overlap and identify consensus molecular clusters from existing SLE signatures ( Figure 1 ). We found five consensus molecular clusters that characterize the heterogeneous SLE transcriptional landscape across existing signatures were: (1) interferon response, (2) lymphoid (T cell, NK cell), (3) plasma cell, (4) neutrophil, and (5) platelet/erythrocyte. We found that autologous anti-CD19 CAR-T cell therapy suppressed interferon, plasma cell, and neutrophil modules, while lymphoid modules were upregulated post CAR-T cell therapy ( Figure 2A ). We next sought to determine whether the transcriptomic “resetting” effects we observed following autologous CAR-T therapy extends to allogeneic approaches. The allogeneic YTS109 CD19-STAR T cells also suppressed interferon, plasma cell, and neutrophil modules, with lymphocyte modules upregulated post CAR-T cell therapy at the month 3 primary endpoint relative to baseline ( Figure 2B ). We found that both the expression and proportion of immature neutrophils, which have been associated with active LN, decreased over time post-allogeneic therapy, with corresponding declines in plasma neutrophil granule protein levels including AZU1, MPO, BPI, and LCN2 (p-trend <0.05) ( Figure 2C ).
Conclusions: Autologous and allogeneic CD19-targeted CAR-T cell therapies induce transcriptional resets in core SLE modules. As we further investigate the molecular profiles of autoimmune patients post CAR-T therapy with larger cohorts, it offers a unique opportunity to identify key pathways to restore normal immune function and assess the durability of immunologic reset.
Consensus SLE molecular signatures reveal core immunological axes. (A) We co-normalized 4,638 bulk transcriptome samples from 22 peripheral blood cohorts and applied existing SLE signatures. (B) Consensus molecular endotypes separated in principal component analysis (PCA). (C) Network analysis of scaled scores (Spearman’s correlation >0.3); edge line thickness represents correlation strength.
Autologous and allogeneic CD19 CAR-T cell therapy modulates core SLE modules. (A) Wilhelm cohort single-cell RNA-seq (n=102,331 cells) from autologous CD19 CAR-T therapy comparing each patient’s post- vs. pre-treatment profiles; Hedges’ g effect size. (B) Wang cohort single-cell RNA-seq (n=541,286 cells) following allogeneic CD19 STAR-T cell product infusion comparing month 3 vs. baseline. (C) Immature neutrophil signatures decrease after treatment at the RNA and protein levels.
REFERENCES: [1] Müller F, et al. Nature Medicine. 2026; doi: 10.1038/s41591-025-04185-6.
[2] Wang X, et al. Nature Medicine. 2025; 31(11):3713-3724.
[3] Wilhelm A, et al. JCI Insight. 2024; 9(12):e179433.
Acknowledgments: NIL.
Disclosure of Interests: Evan Maestri: None declared, Holly McCann: None declared, Hong Zheng: None declared, Andreas Kerschbaumer Galapagos, JNJ, MSD, Novartis, Pfizer, Stada, UCB, AbbVie, Lilly, JNJ, UCB, Andrew Moore: None declared, Paul J Utz reported receiving grants from Pfizer., Purvesh Khatri reported receiving grants from Pfizer and grants from Eli Lilly, and is co-founder and consultant to Inflammatix, Inc.