fetching data ... 
Background: Familial Mediterranean fever (FMF) is classically considered an autosomal recessive pyrin-inflammasome autoinflammatory disease. However, simple heterozygous MEFV carriers can present with clinically compatible inflammatory attacks, raising a major unresolved question: to what extent do heterozygous and bi-allelic crises share the same systemic inflammatory biology, and which molecular layers best capture genotype-linked divergence beyond clinical overlap?
Objectives: To define genotype-associated immune programs during FMF acute attacks by comparing bi-allelic versus simple heterozygous patients using a multi-omics framework, and to identify tractable circulating biomarkers reflecting genotype-dosage effects on inflammatory intensity.
Methods: We analyzed an independent ImmunAID cohort of 38 FMF patients sampled during acute attacks (27 bi-allelic and 11 simple heterozygous). Multi-layer profiling included bulk PBMC RNA-seq, serum cytokine profiling, SomaScan plasma proteomics (~8,000 proteins), and flow cytometry immune phenotyping. Differential transcriptomic and proteomic analyses were performed with false discovery rate control, complemented by canonical signaling pathway enrichment and program-level inference prediction. Soluble inflammatory mediators were quantified using a broad targeted panel (>80 analytes) spanning alarmins, cytokines, chemokines, and acute-phase reactants (including CRP and SAA), alongside plasma proteomics and cellular immune profiling.
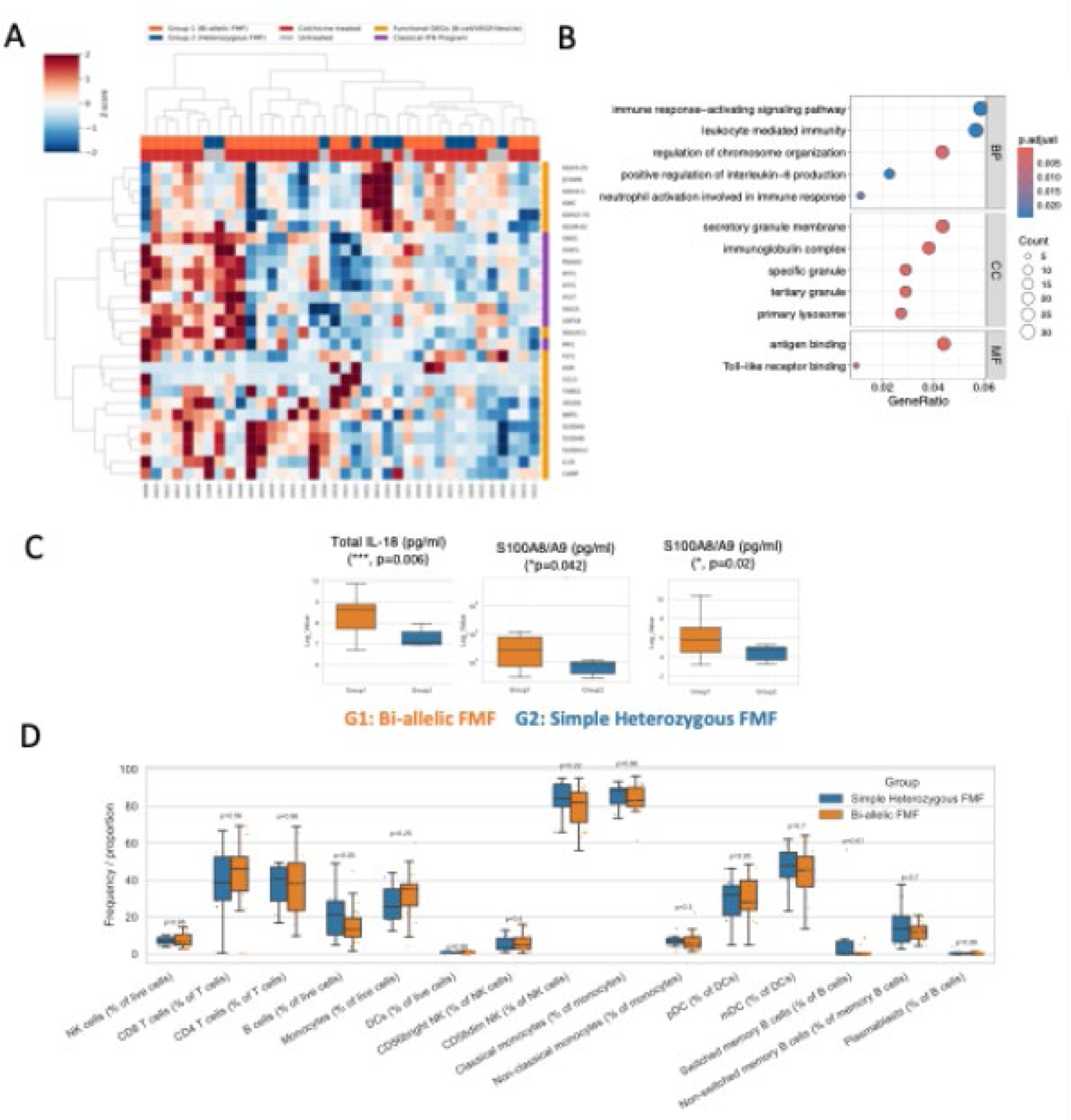
Results: Bulk PBMC RNA-seq identified robust genotype-associated divergence between bi-allelic and simple heterozygous FMF during attacks (23 upregulated and 617 downregulated DEGs). Unexpectedly, the dominant structure was not a canonical innate cytokine module but a strong adaptive/humoral skew, driven by immunoglobulin variable and constant-region transcripts (e.g., IGHV/IGLV/IGKV, JCHAIN, IGKC ) alongside a smaller set of non-Ig contributors ( SIGLEC1, THBS1, NINJ1, METRNL ). GO enrichment reinforced immunoglobulin complex and humoral programs with additional granule/lysosome and neutrophil-linked terms. In contrast, interferon-stimulated genes showed no genotype-defining structure, with only occasional IFN-high outliers in bi-allelic patients. Program inference further revealed a bidirectional shift, with increased VEGF-associated activity (supported by RETN and a plasmablast-like module such as JCHAIN/MZB1 ) and relatively attenuated NF-κB-associated activity in bi-allelic disease (Figure 1). Unbiased plasma SomaScan profiling did not identify proteins differing between genotypes during attacks after multiple-testing correction. However, targeted mediator measurements resolved a genotype-linked inflammatory axis: total IL-18 was higher in bi-allelic patients (p=0.006), together with increased S100A8/A9 (calprotectin) (p=0.042) and S100A12 (p=0.02). Although CRP and SAA were numerically increased in bi-allelic patients, variability limited statistical significance. Flow cytometry showed no significant differences in PBMC lineage frequencies, suggesting transcriptomic divergence reflects functional immune-state differences rather than major compositional shifts (Figure 1).
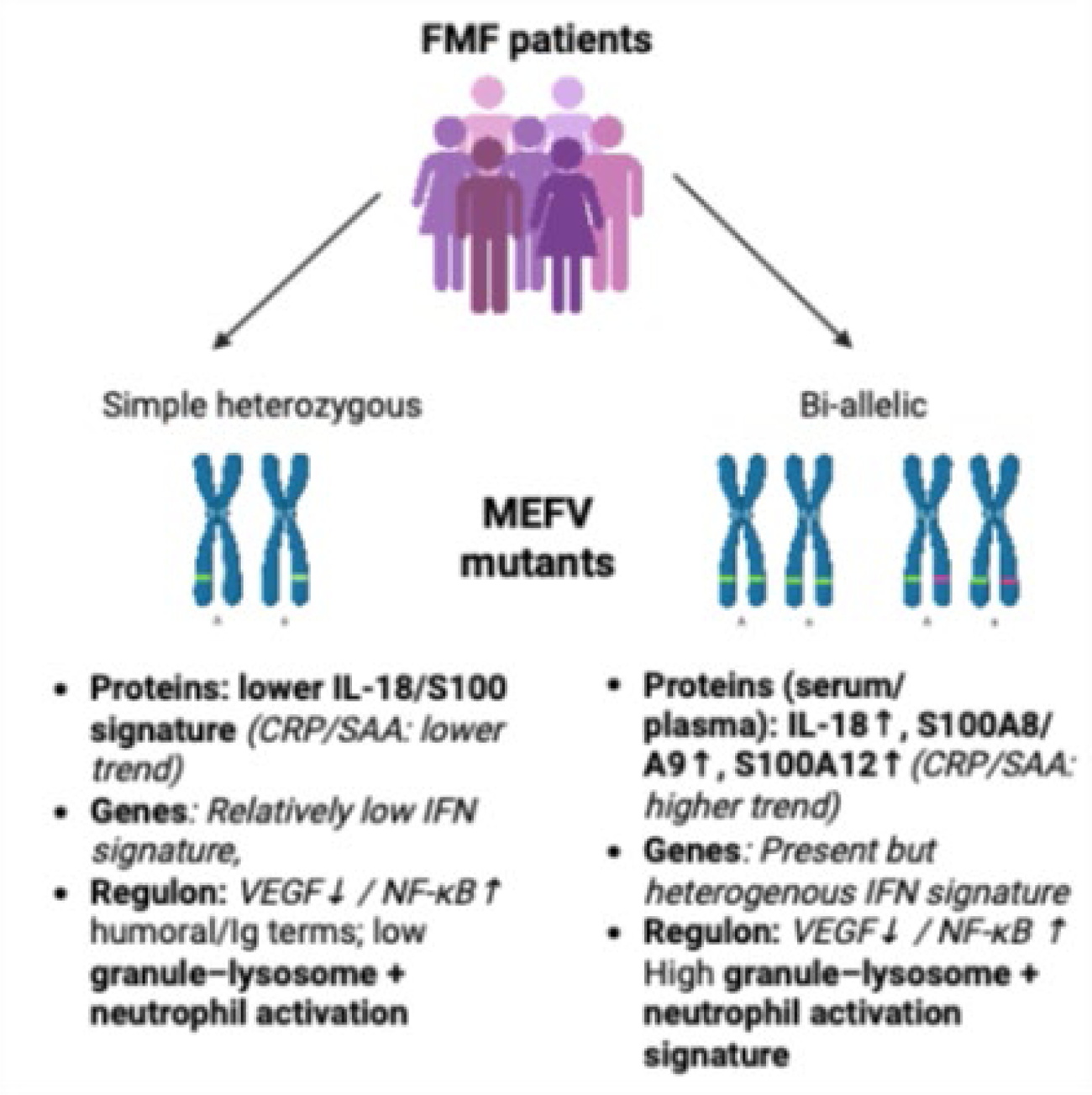
Conclusions: Despite overlapping clinical phenotypes, bi-allelic and simple heterozygous FMF attacks exhibit distinct multi-omic immune programs, characterized by prominent humoral/immunoglobulin-linked PBMC transcriptional divergence and a stronger IL-18/S100 inflammatory axis among bi-allelic patients. These findings support a genotype-dosage effect on systemic inflammatory intensity and nominate IL-18 with S100A8/A9 and S100A12 as tractable biomarkers for FMF stratification and early intervention during attacks (Figure 2).
Distinct immune programs in bi-allelic (hmz) versus simple heterozygous (htz) FMF attacks
(A) PBMC transcriptomic divergence and pathway enrichment by genotype.
Heatmap shows scaled expression of differentially expressed genes between hmz FMF and htz FMF sampled during acute attacks. Genes and samples are hierarchically clustered, revealing a dominant genotype-associated module enriched for immunoglobulin/humoral programs together with immune activation terms.
(B) Gene Ontology enrichment of differentially expressed genes (DEGs) between hmz FMF and htz FMF patients.
Dot plot summarises Gene Ontology enrichment of the genotype-discriminative transcriptome, highlighting over-representation of pathways related to immune activation, granule/lysosome biology, and immunoglobulin complex/antigen binding (bubble size indicates gene count; color indicates adjusted P value).
(C) Soluble inflammatory mediators identify an IL-18/S100 axis in bi-allelic patients.
Boxplots from the targeted soluble mediator panel (>80 analytes) show significantly higher circulating total IL-18, S100A8/A9 (calprotectin) and S100A12 in hmz FMF compared with htz FMF during attacks (P values indicated on plots). These mediators emerge as discriminative markers of genotype-linked inflammatory intensity.
(D) Similar PBMC lineage composition across genotypes during attacks. Flow cytometry quantification of major PBMC populations demonstrates broadly comparable immune cell frequencies between genotype groups, indicating that the observed transcriptomic divergence primarily reflects immune-state differences rather than major shifts in cellular composition.
Genotype-associated multi-omics programs during FMF acute attacks. The schematic integrates: (i) targeted circulating mediators, showing a stronger IL-18/S100A8/A9/S100A12 axis in bi-allelic FMF (with CRP/SAA trends indicated); (ii) PBMC transcriptomic patterns, dominated by immunoglobulin/B cell–linked transcripts with limited interferon-stimulated gene structure; (iii) Gene Ontology enrichment highlighting humoral/Ig-related terms alongside granule–lysosome and neutrophil activation annotations; and (iv) inferred upstream program directionality distinguishing genotype groups, including VEGF and NF-κB regulatory activity signatures.
REFERENCES: NIL.
Acknowledgments: NIL.
Disclosure of Interests: None declared.