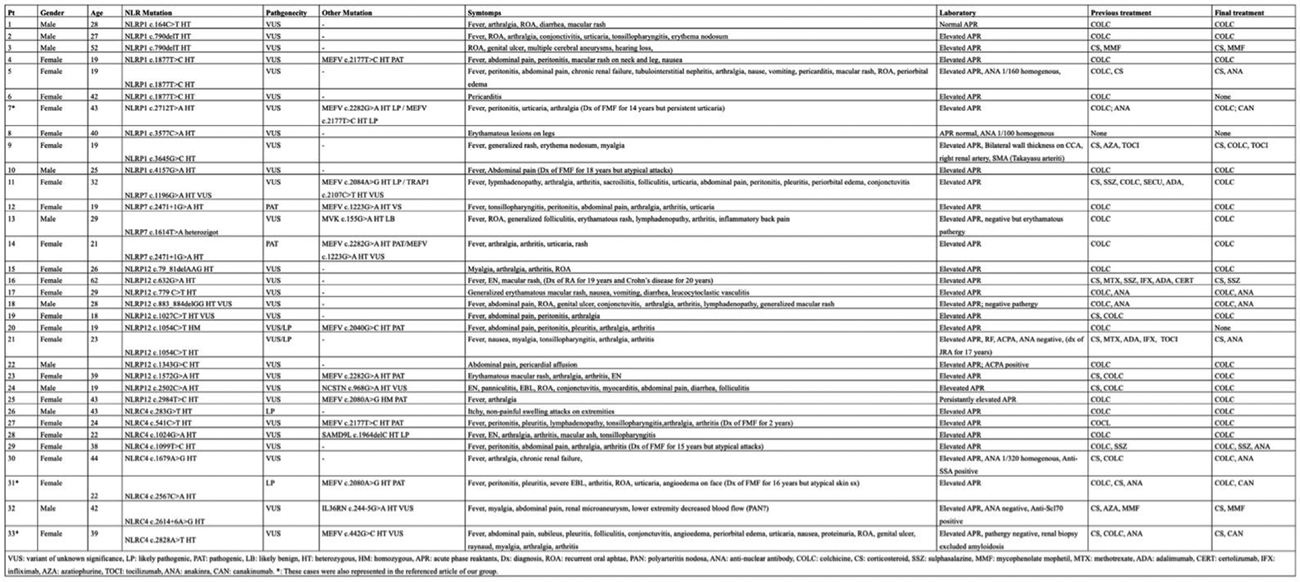
fetching data ... 
Background: Gain-of-function mutations in NLRP3 have long been recognized as the prototypical cause of inflammasome-driven autoinflammatory diseases (AIDs), accumulating evidence indicates that a broader spectrum of related genes contributes to the phenotype. In addition to NLRP3 , other members of the nucleotide-binding domain and leucine-rich repeat–containing (NLR) family, including NLRP1, NLRP7, NLRP12, and NLRC4 , have emerged as important mediators of systemic inflammation with variable clinical expression. Other NLR inflammasome components may be involved in atypical or overlapping inflammatory phenotypes, but the clinical relevance and phenotypic spectrum associated with these variants remain incompletely defined.
Objectives: To characterize the clinical, laboratory, and genetic features of patients carrying variants in NLRP1, NLRP7, NLRP 12 or NLRC4 and to explore their contribution to the spectrum of systemic AIDs (SAIDs).
Methods: We conducted a retrospective analysis of the patients referred for suspected SAIDs who were found to harbor variants in NLRP1, NLRP7, NLRP12, or NLRC4. All patients underwent targeted next-generation sequencing (NGS) using the Ion AmpliSeq Library Kit Plus Autoinflammatory Panel. Genetic variants were interpreted according to American College of Medical Genetics and Genomics (ACMG) criteria (e.g., pathogenic, likely pathogenic, variant of unknown significance, benign, likely benign), and clinical phenotypes were analyzed.
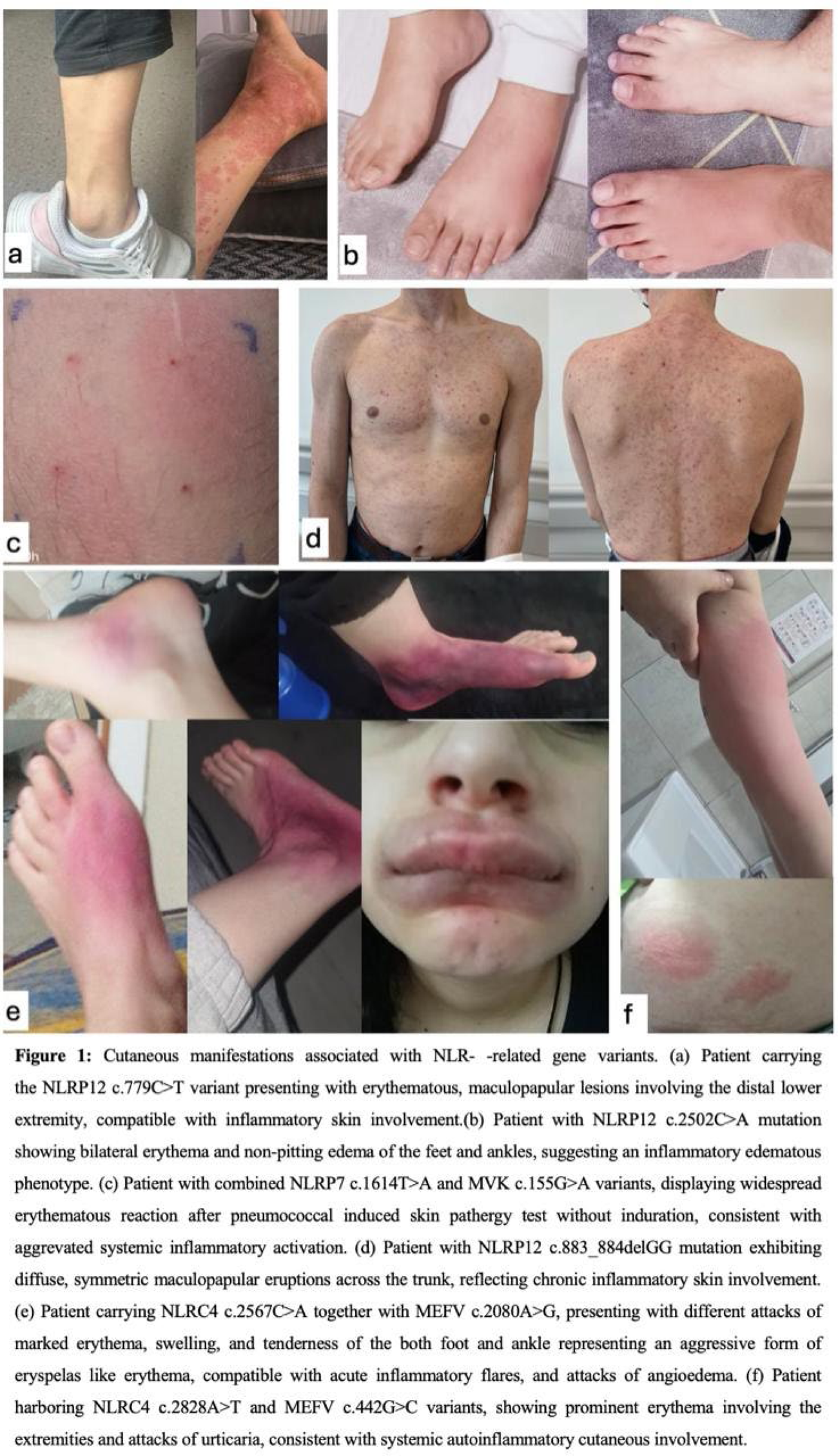
Results: 33 patients aged 19-62, with female predominance (23, 70%) and heterogeneous but overlapping inflammatory phenotypes (Table 1) were included. Recurrent fever was the most common feature, often accompanied by musculoskeletal involvement (arthralgias or episodic arthritis) and prominent mucocutaneous manifestations. Notably, a high prevalence of skin and mucosal findings was observed across the cohort, including erythematous rashes, urticarial lesions, erythema nodosum-like plaques, recurrent oral aphthae, and conjunctivitis (Figure 1). Despite overlapping symptoms, distinct patterns emerged for each NLR gene variant group (Table 1). Patients with NLRP1 mutations often had recurrent fever and serositis reminiscent of FMF, and a subset showed atypical findings such as persistent skin lesions or co-occurring autoimmune findings (e.g., one patient developed large-vessel arteritis, and another had chronic arthritis with positive ANA), which suggests that NLRP1 variants can present with mixed autoinflammatory and autoimmune features. Patients with NLRP7 mutations had recurrent fevers and mild to severe systemic inflammation. Interestingly, one patient with an NLRP7 variant (c.1614T>A) also carried a low-activity MVK variant and presented with recurrent attacks of fever, rash, lymphadenopathy, and an erythematous, non-indurated skin pathergy reaction (Figure 1c). This suggests NLRP7 variants can contribute to a febrile and mucocutaneous dominant auto-inflammatory phenotype in addition to their known role in reproductive disorders. Several patients with NLRP12 mutations experienced recurrent fever with generalized maculopapular skin eruptions or erythema nodosum, suggesting a periodic fever syndrome phenotype. Arthralgia or arthritis, and lower extremity dominant erythematous lesions were common (Figure 1a-b). Patients carrying NLRC4 mutations often had episodic systemic inflammation characterized by high fever and gastrointestinal symptoms, but also experienced recurrent erysipelas-like erythema and episodes of angioedema affecting the face or extremities. These atypical skin findings (Figure 1e-f) are not characteristic of hemophagocytic syndrome, which has been linked to NLRC4, and indicate an inflammasome-driven cutaneous component associated with NLRC4 variants.
Importantly, many patients had coexisting heterozygous mutations in the MEFV but atypical symptoms such as persistent urticaria, severe erythematous flares, or oral apthae in addition to the FMF findings. Our data suggest that these unexpected features were likely attributable to the co-occurring NLR variant, highlighting a possible synergistic effect between the MEFV mutations and individual NLR gene variants in producing a complex clinical picture.
Nearly all patients were initially given colchicine, especially those with FMF-like symptoms. In about one-third of cases, colchicine alone or sometimes combined with low-dose corticosteroids effectively controlled fever and serositis, even in patients without MEFV mutations. Notably, several patients carrying NLRP1, NLRP7, or NLRP12 variants achieved good disease control with colchicine. However, others required a treatment escalation to biologic agents. About one-third required IL-1 blockade (anakinra or canakinumab) to suppress refractory inflammatory flares. Patients with NLRC4 mutations and severe skin involvement often fell into this category of biologic requirement. A few individuals with overlapping autoimmune features were treated with infliximab or tocilizumab, based on their specific diagnoses.
Conclusions: Our cohort expands the understanding of NLR-associated autoinflammatory diseases beyond the well-known NLRP3 variants. We demonstrate that pathogenic or rare variants in NLRP1, NLRP7, NLRP12, and NLRC4 can cause a broad and overlapping spectrum of systemic inflammation, often marked by prominent mucocutaneous involvement. Clinicians should be vigilant for unusual skin manifestations in SAID patients, including unexplained recurrent erythema, urticarial rash, or angioedema, which should guide genetic screening beyond the MEFV. Our findings also highlight that a substantial subset of these non-classical SAID patients may respond to colchicine therapy. Lastly, the NLR variants may co-exist with the MEFV and other autoinflammatory variants and contribute to the mixed autoinflammatory phenotype [1].
Table 1 . Detailed description of each patients with any of NLRP1, NLRP7, NLRP 12 or NLRC4 variants.
REFERENCES: [1] Amikishiyev S, et al. Rheumatology (Oxford). 2025.
Acknowledgments: NIL.
Disclosure of Interests: None declared.