fetching data ... 
Background: Osteoporosis is a common comorbidity across inflammatory diseases, which presents unique challenges in axial spondyloarthritis (axSpA). Abnormal bone formation alters spinal biomechanics which, when combined with reduced bone mineral density, increases susceptibility to potentially catastrophic vertebral fractures. Unlike other inflammatory conditions, the causal direction between osteoporosis and axSpA remains unclear. Baseline osteoporosis is associated with increased structural progression in axSpA, with Mendelian randomization (MR) supporting a osteoporosis-to-axSpA causal direction [1]. However, recent genome-wide association studies (GWAS) identified a strong genetic correlation between radiographic axSpA (hence forth abbreviated as AS) and low bone density and fracture [2]. Such shared genetic architecture may explain both traits (pleiotropy) and generate spurious directional associations using MR [3].
Objectives: To examine whether a causal relationship exists between osteoporosis and AS and, if not, to identify shared genetic mechanisms that may explain the observed associations
Methods: Genetic association data for osteoporosis (20,117 cases; 422,420 controls) were obtained from the Million Veteran Program, restricted to participants of European ancestry. Osteoporosis was defined using Phecode 741.1 (ICD-9 code 733.0* not solely based on bone density). GWAS summary statistics for AS were derived from 8,244 cases meeting modified New York criteria and 14,274 European-ancestry controls [4].
We estimated genome-wide genetic correlation between AS and osteoporosis using linkage disequilibrium score regression (LDSC). Where a significant correlation was observed, we applied a Latent Causal Variable (LCV) model to assess whether the shared genetic component is consistent with a directional causal relationship versus pleiotropy. LCV models a single latent variable mediating the genetic correlation and estimates the genetic causality proportion (GCP); values approaching +1 (osteoporosis → AS) or -1 (AS → osteoporosis) support directional effects, whereas values near zero suggest shared genetic architecture without clear directionality.
To identify specific shared loci, we applied PLACO (pleiotropic analysis under a composite null hypothesis), which tests for joint association with both traits—rather than association with none or only one—using the product of the Z statistics for each trait at the same variant. Variants with minor allele frequency <0.01 and with Z 2 >80 were excluded (to reduce bias from extremely large effects). Sample overlap was estimated using SNPs with P≥1×10 -4 in both traits and accounted for where applicable. Variants surpassing the prespecified pleiotropy threshold (P<5×10 -8 ) were functionally annotated in FUMA. Pleiotropic SNPs were clumped into distinct loci within a ±250 kb radius using LD thresholds r 2 <0.6 for independent significant variants and r 2 <0.1 for lead SNPs (1000 Genomes Phase 3 reference panel). Lead SNPs were mapped to protein-coding genes using positional mapping (≤10 kb).
For each lead locus, we performed Bayesian colocalization using variants within ±200 kb of the lead SNP to assess whether osteoporosis and AS association signals are consistent with a posterior probability (PP) of shared causal variant (H4) versus distinct causal variants (H3). Variants in the extended MHC region (chr6:25-34Mb) were excluded due to complex linkage disequilibrium. We used default priors and considered loci with conditional posterior probability of shared causal variant (PPH4/(PPH3+PPH4)) >0.7 as supporting colocalization.
Results: Osteoporosis and AS demonstrated a significant positive genome-wide genetic correlation (rg=0.29, SE=0.09, P=0.001). However, LCV analysis provided no evidence for a directional causal relationship (GCP=0.09, P=0.97), consistent with substantial shared genetic architecture rather than causality.
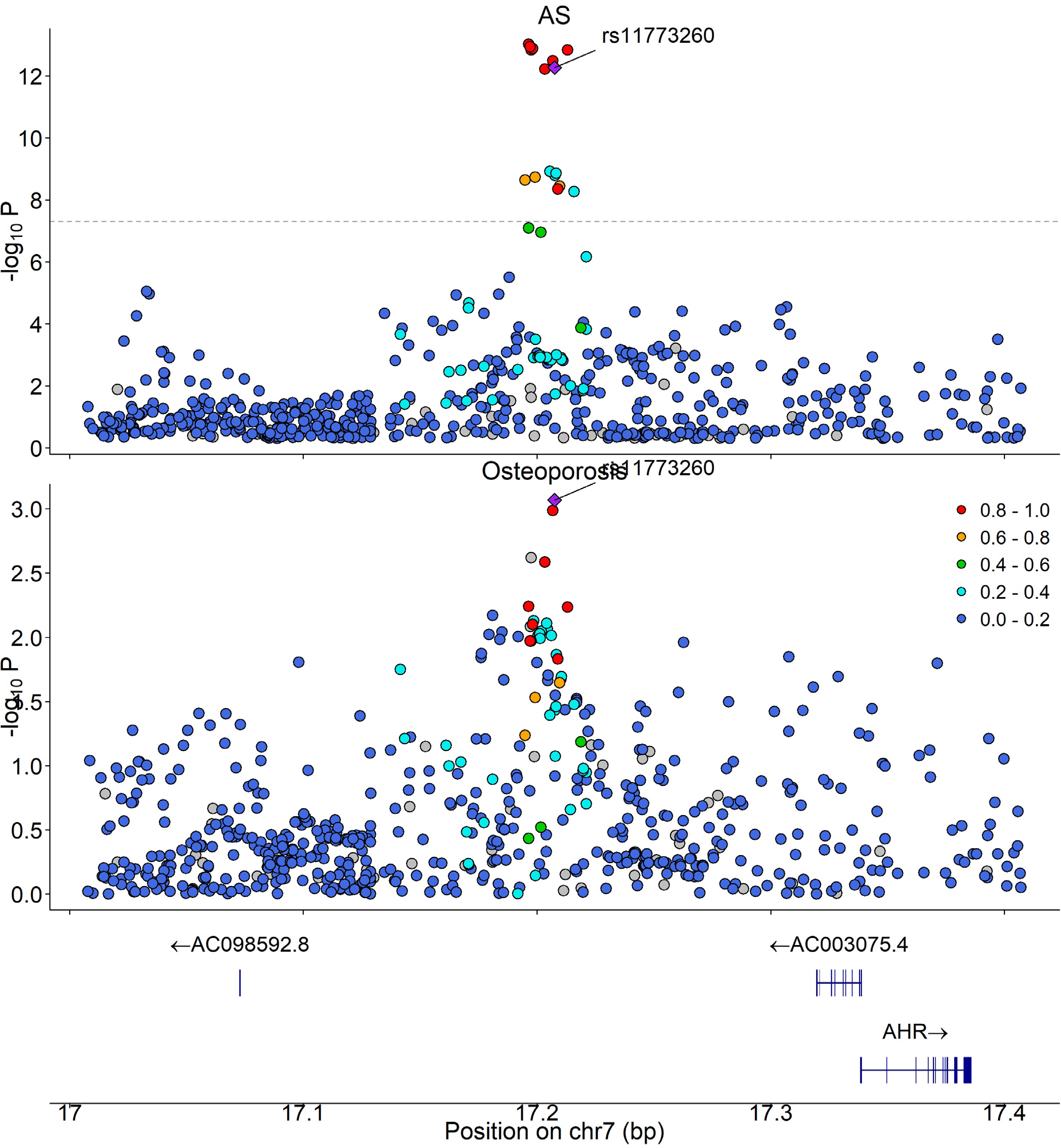
PLACO identified 647 variants jointly associated with osteoporosis and AS, which mapped to six pleiotropic genomic loci after linkage disequilibrium-based clumping (Table 1). Following exclusion of loci within the extended MHC region, four lead (index) SNPs were taken forward for colocalization analysis, of which one locus on 7p21.1 (lead SNP rs11773260), proximal to AHR , showed evidence consistent with a shared causal variant (Figure 1).
Conclusions: Osteoporosis and AS share substantial genetic architecture but show no evidence of a directional causal relationship, suggesting that previously reported MR associations are unlikely to be explained by genetic liability to one condition causally driving the other. Genome-wide pleiotropy analysis identified a limited number of shared loci, with robust evidence for colocalization at a single locus on 7p21.1 near AHR , encoding the aryl hydrocarbon receptor. AHR is a transcription factor that links immune regulation, including critically activation of Th17 and other IL-17-producing lymphocytes, to bone remodelling, increasing osteoclast activation and suppressing osteoblast differentiation. While colocalization analyses may be underpowered at loci with modest effects or complex linkage disequilibrium, these findings refine the genetic basis of osteoporosis in AS and highlight specific pathways for targeted functional and translational investigation.
Lead SNPs at pleiotropic loci jointly associated with osteoporosis and AS.
| Locus | Lead SNP | Chr:Pos (hg19) | P (PLACO) | Nearest gene |
|---|---|---|---|---|
| 1 | rs6665959 | 1:67,728,816 | 8.91×10 -9 | IL23R region |
| 2 | rs9651055 | 1:154,360,684 | 5.84×10 -9 | IL6R/ATP8B2 region |
| 3 | rs3767502 | 1:201,023,098 | 3.45×10 -8 | INAVA region |
| 4 | rs1165206; rs1971508 | 6:25,867,566; 6:26,085,807 | 5.42×10 -9 ; 1.02×10 -10 | MHC region |
| 5 | rs35814746 | 6:28,299,088 | 4.78×10 -9 | MHC region |
| 6 | rs11773260 | 7:17,207,578 | 2.81×10 -8 | Near AHR |
Colocalization of genetic association signals for osteoporosis and AS at the 7p21.1 locus
REFERENCES: [1] Mei J, et al. Front Immunol. 2023;14:1163258.
[2] Brown M, et al. Res Sq. 2025;rs.3.rs-6917334.
[3] O’Connor LJ, Price AL. Nat Genet. 2018;50:1728–34.
[4] Li Z, et al. Ann Rheum Dis. 2021;80:1168–74.
Acknowledgments: NIL.
Disclosure of Interests: None declared.