fetching data ... 
Background: Rheumatoid arthritis (RA) is a chronic autoimmune disease that can lead to irreversible joint damage and disability without timely, effective treatment. Treat-to-target strategies aim for remission, yet current outcome measures rely heavily on subjective clinical assessments and non-specific acute-phase reactants. This lack of objective metrics also confounds clinical trials, resulting in enrollment of patients with incorrect diagnoses or inactive disease [1]. There is an unmet need for an objective, broadly generalizable biomarker that supports RA classification and tracks disease activity to improve trial design and care.
Objectives: To develop and validate a peripheral blood transcriptomic signature that distinguishes RA from healthy controls and correlates with objective disease activity using the largest multi-cohort transcriptomic dataset in patients with RA.
Methods: We systematically searched the Gene Expression Omnibus and ArrayExpress for peripheral blood transcriptomic datasets in RA, including microarray and bulk RNA-sequencing of whole blood or PBMCs. We only included datasets comprising at least 10 samples RA patients and healthy controls (HC). Wherever available, we extracted data on disease activity assessments (CDAI, SDAI, DAS28).
Using bayesMetaIntegrator, we identified genes consistently dysregulated in RA versus HC across discovery studies (absolute effect size ≥0.525; posterior probability ≤0.05). We quantified classification performance by the area under the receiver operating characteristic curve (AUC) using the difference in the geometric mean of up- and downregulated genes as a classifier and validated in independent cohorts.
We co-normalized all datasets using COCONUT (COmbat CO-Normalization Using conTrols) to enable integrated analyses with phenotype data. Unsupervised disease trajectory inference was used to refine the signature to genes associated with disease activity and to group them into modules. Public single-cell RNA-seq datasets (Binvignat et al. JCI Insight 2024, n=36, 98335 cells; He et al. Sci Transl Med 2025; n=53, 709460 cells) were leveraged to infer cell-type origins [2,3].
We evaluated the associations with objective disease activity using a separate, large observational study on DMARD naïve patients (RA-MAP, GSE97810; n=187) [4]. To evaluate the correlation of the signature with core set measures we used linear regression (swollen joint count [SJC], tender joint count [TJC], patient global assessment [PGA], evaluator global assessment [EGA], C-reactive protein [CRP]) and the Simplified Disease Activity Indes (SDAI).
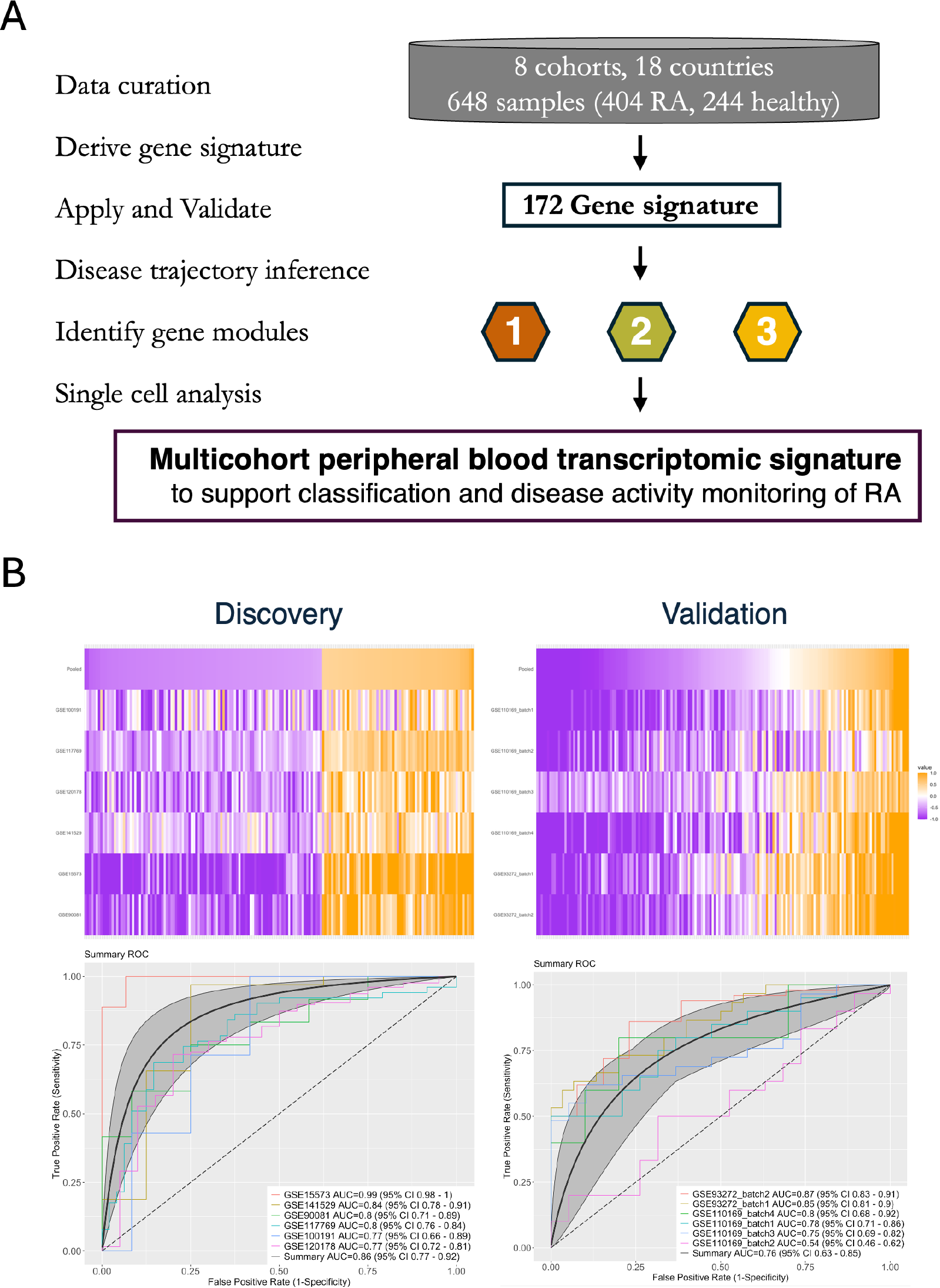
Results: We curated 8 cohorts from 18 countries, totaling 404 RA patients and 244 healthy controls with peripheral blood transcriptomic data. The discovery set comprised 6 cohorts (4 bulk RNA-seq, 2 microarray; 247 RA, 117 HC); the validation set comprised 2 microarray cohorts (6 batches; 157 RA, 127 HC).
Using BayesMetaIntegrator we identified a 172-gene signature (67 upregulated, 105 downregulated) with an AUC of 0.86 (95% CI 0.77-0.92) in the discovery set and 0.76 (95% CI 0.63-0.85) in validation (Figure 1, Panel B). After co-normalization, we used trajectory inference to reduce the gene signature to 40 key genes, divided into three distinct modules with correlated expression. When conducting single-cell analysis utilizing 2 separate datasets (807795 cells), we found that these three modules had distinct cellular pathways: Module 1 (13 genes) was predominantly expressed in lymphocytes and plasmablast, and inversely associated with disease activity; Module 2 (14 genes) was neutrophil-enriched and most discriminative for RA versus HC; Module 3 (13 genes) was enriched for neutrophil and monocytes, and most strongly associated with disease activity.
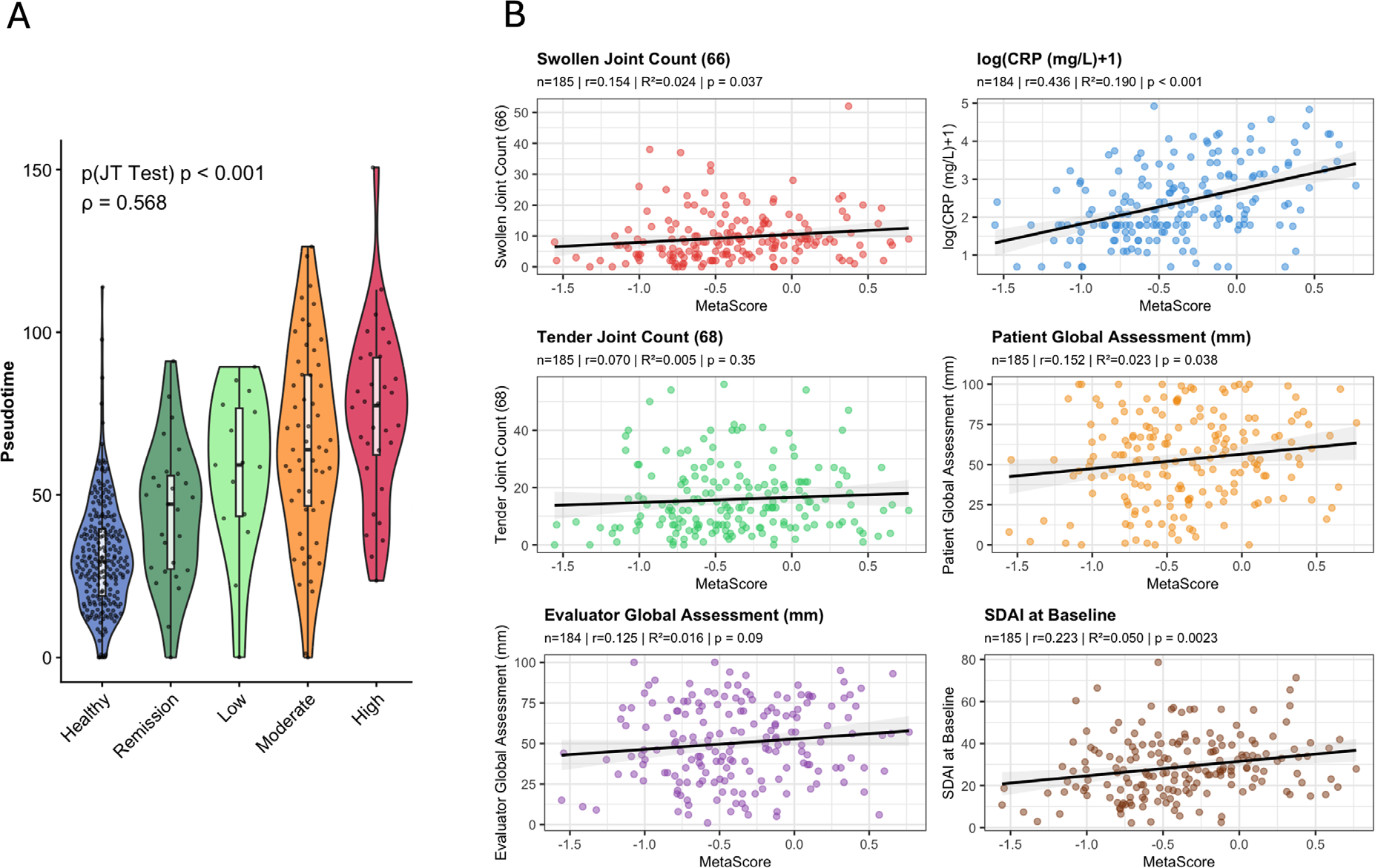
We could also show that, beyond being a diagnostic signature, this refined 40-gene signature was highly associated with disease activity (Spearman rho=0.568, p<0.001; Figure 2, Panel A). In an independent cohort of 187 DMARD naïve RA patients (RA-MAP, GSE97810), we identified a correlation of the 40-gene score with SJC (r=0.154, p=0.037), CRP (r=0.436, p<0.001), SDAI (r=0.223, p=0.002), and PGA (r=0.152, p=0.038), but not with TJC (p=0.35) or EGA (p=0.09) in linear regression analyses (Figure 2B).
Conclusions: To our knowledge, this is the largest multicohort analysis of peripheral blood transcriptomics in RA that is representative of biological and clinical heterogeneity of real-world patient population. The 40-signature robustly distinguishes RA from HC and tracks disease activity, despite the biological and clinical heterogeneity across datasets. This signature shows the strongest correlations with objective measures of disease activity (SJC, CRP) and SDAI and reflects distinct cellular processes, with protective lymphocyte signals and pro-inflammatory myeloid signals. It has potential as an objective biomarker to support RA classification and disease activity monitoring and may enhance patient selection and endpoint assessment in clinical trials.
Peripheral blood gene expression data from 8 cohorts, 648 samples (rheumatoid arthritis n=404, Healthy control n=244) co-normalized. Panel A: Methodology to derive a multicohort peripheral blood transcriptomic signature. Panel B: 172 gene signature is diagnostic for rheumatoid arthritis and validates in separate datasets.
Trajectory analysis identified a subset of 40 relevant genes that are associated with disease activity. Panel A: Association of pseudotime with disease activity in the COCONUT co-normalized discovery dataset (n=648). Panel B: Validation of the 40 gene signature with individual core set measures of disease activity in an independent dataset of treatment naïve patients with rheumatoid arthritis (n=185).
REFERENCES: [1] Kerschbaumer A, et al. Ann Rheum Dis 2025;84:1632–40.
[2] Binvignat M et al. JCI Insight 2024;9.
[3] He Z et al. Sci Transl Med 2025;17:eadt7214.
[4] RA-MAP Consortium Sci Data 2022;9:196.
Acknowledgments: NIL.
Disclosure of Interests: Andreas Kerschbaumer Galapagos, JNJ, MSD, Novartis, Pfizer, Stada, UCB, AbbVie, Lilly, JNJ, UCB, Andrew Moore: None declared, Evan Maestri: None declared, Holly McCann: None declared, Ananthakrishnan Ganesan: None declared, Hong Zheng: None declared, William Robinson Ebvio Inc, Flatiron Bio LLC, Purvesh Khatri Co-founder of Inflammatix, Inflammatix.