fetching data ... 
Background: The current ACR/EULAR classification criteria recognize four subsets of inflammatory myopathies (IM): dermatomyositis (DM), immune-mediated necrotizing myopathy (IMNM), inclusion body myositis (IBM), and antisynthetase syndrome (ASyS) [1,2]. We and others have identified scleromyositis (SM) as a fifth subset that differs from the above by the presence of scleroderma features (fulfilling or not the 2013 ACR/EULAR criteria for systemic sclerosis (SSc)), SSc-specific or overlap autoantibodies (most frequently anti-PM/Scl, -U1-RNP, and -Ku), and a distinctive muscular capillaropathy [3]. These patients are often misclassified as seronegative IMNM or ASyS using current ACR/EULAR myositis criteria.
Objectives: This study aimed to test the hypothesis that SM also differs from other IM subsets by a distinct muscle transcriptomic signature.
Methods: In a bicentric cohort (Strasbourg–Montreal), 54 untreated patients with IM at the time of diagnosis were included (SM=23, DM=10, IMNM=8, ASyS=9, IBM=4), along with 7 subjects without neuromuscular disease (controls). Clinico-sero-pathological data were collected at diagnosis and at last follow-up. Transcriptomic sequencing was performed using total RNA extracted from diagnostic muscle biopsies, and pathway enrichment analysis was conducted on differentially expressed genes (|logFC|>0.5; adjusted p<0.05) using WebGestalt 2024.
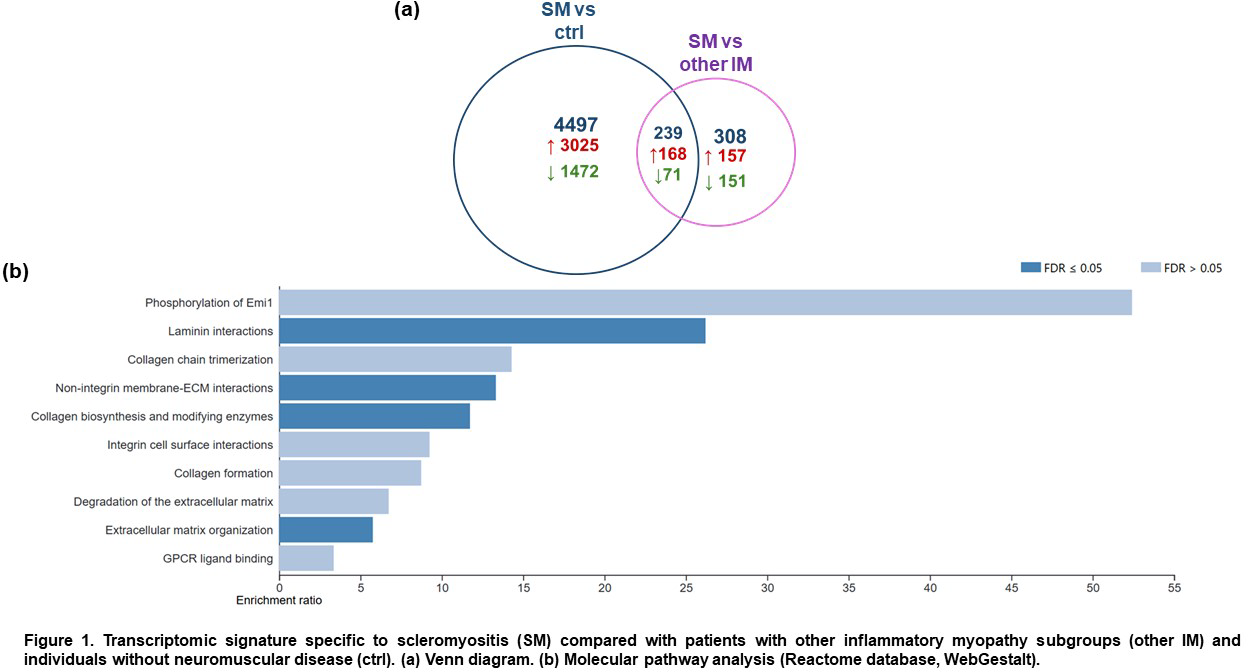
Results: Subjects in each subset were comparable regarding age at diagnosis, sex ratio, muscle biopsy site, and sample origin (p>0.05). Compared to other IM subsets, SM patients exhibited higher frequencies of sclerodactyly (62%, p<0.0001), Raynaud’s phenomenon (61%, p=0.002), limited cutaneous sclerosis (57%, p=0.0006), telangiectasias (52%, p=0.003), interstitial lung disease (47.6%, p=0.03), puffy fingers (38%, p=0.02), and arthritis (38%, p=0.07). The SM cohort encompassed a broad serological spectrum of the disease, including 4 anti-PM/Scl+, 5 anti-U1-RNP+, 4 anti-Ku+, 2 anti-RNA Pol III+, 1 anti-NVL+, 1 anti-Scl70+, 1 anti-RuvBL1/2+, and 5 seronegative patients. A total of 239 genes were specifically dysregulated in SM muscle compared with the four other IM subsets and controls (168 upregulated, 71 downregulated) (Figure 1a). These genes were enriched in pathways related to collagen formation and extracellular matrix remodelling (Figure 1b).
Conclusions: Consistent with their distinctive clinical, serological, and histopathological features, SM patients differ from other IM subsets by enhanced activation of fibrotic molecular pathways in muscle tissue. These findings further support the hypothesis that SM represents a distinct IM subset that should be recognized in future IM classification criteria, and that myositis constitutes a target organ involvement in SSc. Muscle fibrosis activation may represent both a diagnostic biomarker and a potential therapeutic target for SM.
REFERENCES: [1] Lundberg IE. Ann Rheum Dis. 2017 Dec;76(12):1955-1964.
[2] Zanframundo G. Ann Rheum Dis. 2025 Jul;84(7):1207-1220.
[3] Ellezam B. Neuropathol Appl Neurobiol. 2022 Dec;48(7):e12840.
Acknowledgments: NIL.
Disclosure of Interests: None declared.