fetching data ... 
Background: Giant cell arteritis (GCA) represents a critical failure of immune resolution, characterized by persistent vascular inflammation [1] despite the paradoxical absence of a Type I Interferon response in the majority of GCA patients [2]. While current therapies focus on broad suppression, they fail to address the underlying molecular checkpoints that govern disease chronicity and relapse. The pathophysiological roles of the acute phase reactant serum amyloid A and its receptor, formyl peptide receptor 2 (FPR2) [3], have been implicated in this process, yet the distal effectors of resolution failure remain undefined.
Objectives: This study aimed to identify the molecular drivers of resolution failure in GCA and to characterize the epitranscriptomic landscape that dictates the transition from active inflammation to a hyper-primed remission state.
Methods: We performed bulk ribonucleic acid (RNA)-sequencing, label free proteomics, and immunophenotyping on peripheral immune cells from newly diagnosed patients, in glucocorticoid-free remission patients, and healthy controls. Mechanisms were dissected using in vitro stimulation with recombinant serum amyloid A and a specific FPR2 agonist (ACT-389949). Transcriptional regulation of the IL-23/STAT3/SOCS3 axis was quantified by quantitative-PCR, and functional cytokine output was measured by multiplex bead-based immunoassay.
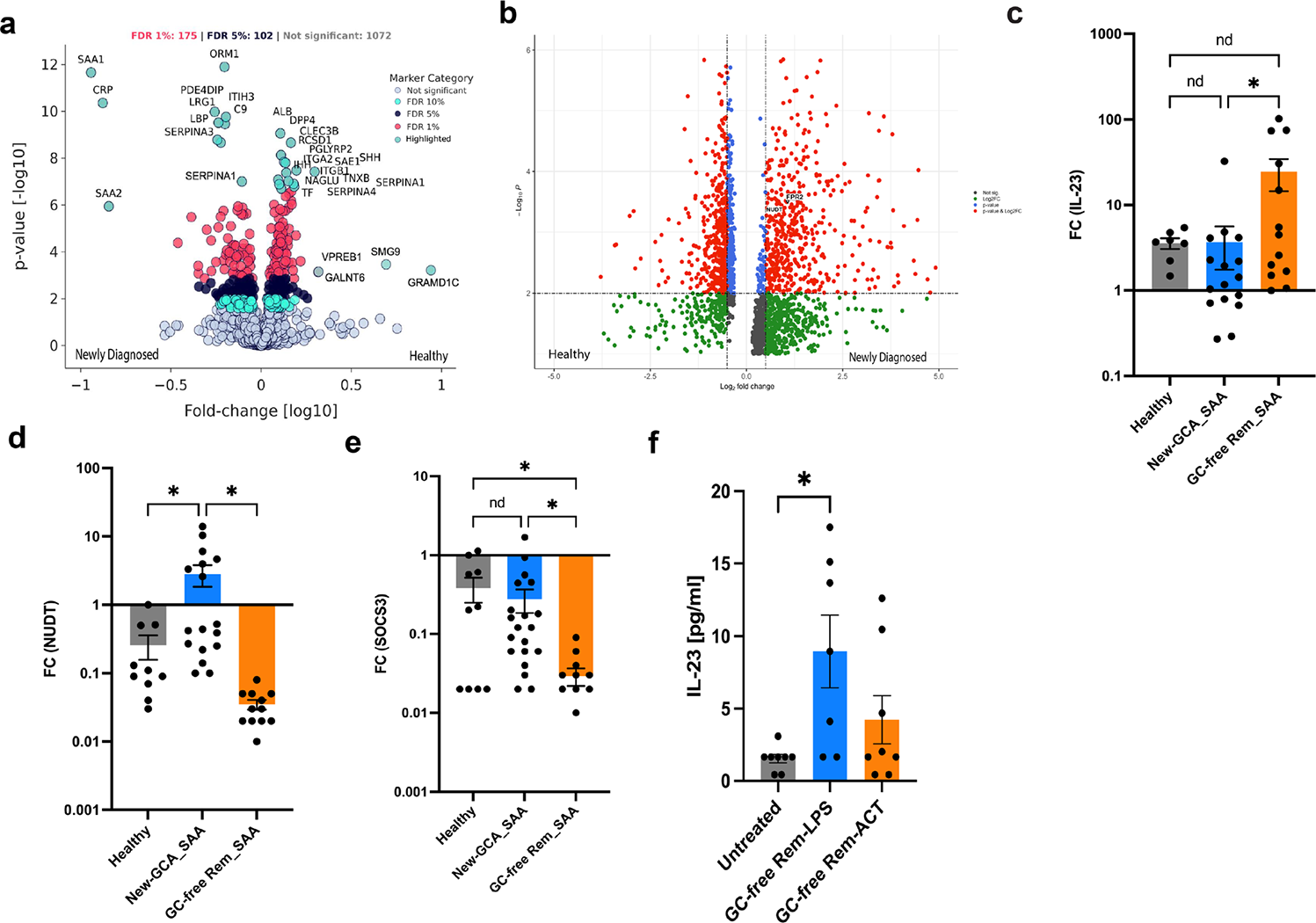
Results: We recruited 31 treatment naïve newly diagnosed GCA patients, 26 patients in glucocorticoid-free remission, and 18 healthy controls at the Division of Rheumatology and clinical immunology, University Hospital Bonn, Germany. High-resolution proteomics identified a systemic ligand switch, where serum amyloid A -1 and -2 dominates active disease state ( Figure 1a ). Active disease is defined by an inflammatory shunt driven by the GTPase-driven RNA decapping enzyme Nudix Hydrolase 16 [4] ( Figure 1b ), which was significantly upregulated and positively correlated with FPR2 expression (r=0.79, p = 0.02). This shunt dismantles 5’-triphosphate-capped RNA, silencing antiviral sensors such as Retinoic Acid-Inducible Gene I while maintaining a controlled burn of Interleukin 23 production ( Figure 1c ). Conversely, remission patients exhibited a hyper-primed state ( Figure 1c ) defined by the collapse of the Nudix Hydrolase 16 shunt ( Figure 1d ) and a failure to induce the suppressor of cytokine signaling 3 ( Figure 1e ). Upon serum amyloid A re-exposure, these brake-deficient cells displayed an explosive, 32-fold upregulation of Interleukin 23 compared to a modest 10-fold increase in active disease ( Figure 1d ). Treatment with the FPR 2 agonist ACT-389949 successfully suppressed inflammation in remission but failed in the refractory active disease state ( Figure 1f ).
Conclusions: These findings identify GTPase RNA decapping as a critical metabolic gatekeeper in GCA. The Nudix Hydrolase 16/suppressor of cytokine signaling 3 axis defines a molecular rev-limiter that maintains chronic inflammation while rendering the remission state molecularly vulnerable to relapse. Targeting this epitranscriptomic shunt offers a novel therapeutic strategy for achieving durable, resolution-based remission in vascular inflammation.
The Serum Amyloid A-Formyl Peptide Receptor 2 Axis and the Nudix Hydrolase 16 Inflammatory Shunt in Giant Cell Arteritis. (a ) Differential expression of serum amyloid A isoforms in patient serum across disease phases. (b ) Transcriptional upregulation of the resolution receptor formyl peptide receptor 2 and the decapping enzyme Nudix Hydrolase 16 in active disease. (c ) Paradoxical Interleukin 23 fold change induction following serum amyloid A stimulation, demonstrating the ceiling effect in active disease and hyper-priming in remission. (d ) Targeted induction of Nudix Hydrolase 16 in newly diagnosed patients following ex vivo serum amyloid A exposure. (e ) Defective induction of the suppressor of cytokine signaling 3 in remission cohort. (f ) Impact of the formyl peptide receptor 2 agonist ACT-389949 on Interleukin 23 protein secretion in remission state.
REFERENCES: [1] Bilton EJ, Mollan SP. Giant cell arteritis: reviewing the advancing diagnostics and management. Eye. 2023 Aug;37(12):2365–73.
[2] Vieira M, Régnier P, Maciejewski-Duval A, Le Joncour A, Darasse-Jèze G, Rosenzwajg M, et al. Interferon signature in giant cell arteritis aortitis. J Autoimmun. 2022 Feb;127:102796.
[3] Mastromarino M, Lacivita E, Colabufo NA, Leopoldo M. G-Protein Coupled Receptors Involved in the Resolution of Inflammation: Ligands and Therapeutic Perspectives. Mini-Rev Med Chem. 2021 Jan 14;20(20):2090–103.
[4] Lu G, Zhang J, Li Y, Li Z, Zhang N, Xu X, et al. hNUDT16: a universal decapping enzyme for small nucleolar RNA and cytoplasmic mRNA. Protein Cell. 2011 Jan;2(1):64–73.
Acknowledgments: NIL.
Disclosure of Interests: None declared.